MERLIN Tutorial -- Modeling Marker-Marker Linkage Disequilibrium
This tutorial describe the procedures and options available for
modeling marker-marker linkage disequilibrium with MERLIN. It assumes that
you are relatively familiar with MERLIN and its standard command line options.
If you haven't yet done so, it is a good idea to first learn about
input file formats and
non-parametric linkage analysis.
MERLIN can accomodate marker-marker linkage disequilibrium in nearly
all available analyses, including parametric and non-parametric analysis
of discrete traits, regression and variance-components based analysis of
quantitative traits, haplotyping analyses and simulation. Modeling marker-marker
linkage disequilibrium is especially important when analysing SNP linkage
maps in datasets where some parental genotypes are missing. It has been
shown that in these settings ignoring marker-marker linkage disequilibrium
can result in severe biases in linkage calculations.
To model linkage disequilibrium, MERLIN organizes markers into clusters.
Each cluster can include both SNP and microsatellite markers. MERLIN then
uses population haplotype frequencies to assume linkage disequilibrium
within each cluster. Two limitations of the model are that it assumes
no recombination within clusters and no linkage disequilibrium between
clusters. These approximations appear to be reasonable in many datasets.
For this example, we will use a simulated data set that you will find
in the examples subdirectory of the MERLIN distribution or in the
download page.
The dataset consists of a SNP linkage scan of a candidate chromosome
in a set of 500 affected sibships, each with three genotyped affected
siblings and one affected parent. The SNP data consists of clusters
of 2-3 SNPs, all within 100kb of each other, genotyped approximately
5cM apart along a single chromosome (20 clusters and 59 SNPs in total).
The three standard Merlin format input files
are the data file snp-scan.dat, pedigree file snp-scan.ped and map file
snp-scan.map. In the pedigree file, SNP alleles 'A', 'C', 'G' and 'T' have
been coded as allele 1, 2, 3 and 4, respectively. All input files are text files,
and you can check their contents using the UNIX more command or using the
following pedstats command:
prompt> pedstats -d snp-scan.dat -p snp-scan.ped
We are going to evaluate the evidence for linkage is this SNP data set,
and a good place to start, is to run a standard non-parametric linkage analysis
(--npl command option) ignoring linkage disequilibrium between markers.
We will request that Merlin carry out analysis at positions spaced every 2 cM
along the chromosome(--grid 2 command line option). Try running
the following command:
prompt> merlin -d snp-scan.dat -p snp-scan.ped -m snp-scan.map --npl --grid 2
After the opening banner screen, your results should be similar to the
following:
Phenotype: DISEASE [ALL] (500 families)
======================================================
Pos Zmean pvalue delta LOD pvalue
min -18.26 1.0 -0.408 -62.47 1.0
max 54.77 0.00000 1.225 301.0 0.00000
0.000 1.28 0.10 0.094 0.57 0.05
5.000 1.85 0.03 0.109 0.96 0.02
10.000 2.38 0.009 0.139 1.56 0.004
15.000 3.23 0.0006 0.204 3.08 0.00008
20.000 4.72 0.00000 0.308 6.75 0.00000
25.000 3.97 0.00004 0.241 4.48 0.00000
30.000 3.62 0.00014 0.234 3.98 0.00001
35.000 2.64 0.004 0.149 1.86 0.002
40.000 2.05 0.02 0.122 1.18 0.010
45.000 2.27 0.012 0.126 1.35 0.006
50.000 1.99 0.02 0.110 1.03 0.015
55.000 2.27 0.012 0.133 1.43 0.005
60.000 2.28 0.011 0.124 1.33 0.007
65.000 3.21 0.0007 0.189 2.84 0.00015
70.000 4.20 0.00001 0.236 4.62 0.00000
75.000 3.78 0.00008 0.220 3.90 0.00001
80.000 2.74 0.003 0.161 2.08 0.0010
85.000 2.23 0.013 0.127 1.34 0.006
90.000 2.20 0.014 0.127 1.33 0.007
95.000 2.28 0.011 0.154 1.66 0.003
100.000 1.87 0.03 0.187 1.65 0.003
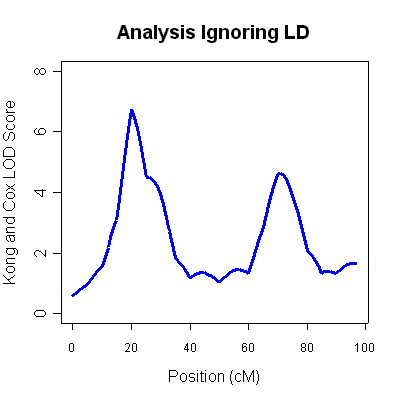
The 4th column (labeled LOD score) is the Kong and Cox LOD score for this
data. You will notice two very strong LOD score peaks, one around 20cM (LOD 6.75) and
another around 70cM (LOD of 4.62). Unfortunately, ignoring marker-marker LD
can lead to inflated LOD scores when some parental genotypes are missing. The results
are typical of
situations where marker-marker disequilibrium is not modeled appropriately and
should not be taken as evidence for linkage.
To verify whether there is evidence for linkage, we will repeat the previous
analysis, but modeling marker-marker disequibrium. First, we will carry out
analyses using pre-specified clusters and haplotype frequencies. Next, we will
see how Merlin can automatically define clusters using the available marker map
and genotype data.
We will use cluster definitions in the file snp-scan.clusters.
This file describes clusters of SNPs in linkage disequilibrium.
This file can be generated by the user, or by a previous MERLIN run.
We will describe the file in detail, since it should help clarify how
MERLIN models linkage disequilibrium.
The file describes a series of clusters, each consisting of a series of
consecutive markers. The description of each cluster begins on a separate
line with the word CLUSTER followed by a series of marker names,
that must exactly match the data and map files. Optionally, this line can
be followed by a series of entries, each on a separate line, describing the
haplotypes in the cluster and their frequencies. Each of these lines begins
with the word HAPLO followed by a haplotype frequency and a series
of alleles.
For example, this is the first cluster in the snp-scan.clusters file:
CLUSTER rs556990 rs553316 rs7989953
HAPLO 0.4500 3 2 1
HAPLO 0.3167 3 2 3
HAPLO 0.2000 1 4 1
HAPLO 0.0333 1 4 3
The cluster includes three markers (rs556990, rs553316, rs7989953) organized
into 4 distinct haplotypes. The first two markers are in complete linkage disequilibrium,
such that allele 3 at rs556990 always appears with allele 2 at rs553316, whereas allele
1 at rs556990 always appears with allele 4 at rs553316. The last marker is in strong, but incomplete
disequilibrium with the first two: allele 3 for rs7989953 nearly always occurs on
the 3-2 haplotype for markers rs556990 and rs553316.
After reading the file with clustering information, MERLIN will do the following:
- Check that all markers within a cluster are contiguous. If they are not, you
will get an error message.
- Check that all markers within a cluster map to the same genetic map position.
If they do not, Merlin will nudge their positions to ensure the within cluster
recombination rate is zero.
- If haplotype frequency estimates are not provided, they will be calculated using
the available genotype data and a maximum-likelihood E-M algorithm.
So let's repeat the original analysis, but modeling of marker-marker disequilibrium
enabled. To do this, use the following command-line:
prompt> merlin -d snp-scan.dat -p snp-scan.ped -m snp-scan.map --npl --grid 5 --cluster snp-scan.clusters
After the opening banner screen, you will first see a series of information messages:
MARKER CLUSTERS: Marker map changed, see [merlin-clusters.log]
MARKER CLUSTERS: User supplied file defines 20 clusters
Family: 101 - Founders: 2 - Descendants: 3 - Bits: 4
Cluster at marker rs7334521 dropped [OBLIGATE RECOMBINANT]
Family: 287 - Founders: 2 - Descendants: 3 - Bits: 4
Cluster at marker rs7334521 dropped [UNKNOWN HAPLOTYPE]
The first two lines indicate that the cluster information was
successfully loaded. Since MERLIN assumes no recombination within
clusters, the original genetic map was adjusted slightly -- you
can examine the contents of merlin-clusters.log for
details. In addition, MERLIN encountered two families (101 and 287)
where genotypes for one cluster did not fit with the model
described in the clustering file. In family 101, the observed
genotypes imply an obligate recombinant in the cluster including
markers rs7334521, rs4495999 and rs9546406. In family 287, the observed
genotypes imply a haplotype that is not present in the clustering file.
In both
families, genotypes for markers rs7334521, rs4495999 and rs9546406
will be marked as missing to allow analysis to proceed. In our experience,
discarding a small proportion of the available genotypes in this manner
results in no noticeable biases.
After these messages, you will find the linkage analysis results, which
should be similar to the following:

Phenotype: DISEASE [ALL] (500 families)
======================================================
Pos Zmean pvalue delta LOD pvalue
min -18.26 1.0 -0.408 -62.47 1.0
max 54.77 0.00000 1.225 301.0 0.00000
0.011 0.82 0.2 0.061 0.24 0.15
5.011 1.17 0.12 0.070 0.39 0.09
10.011 1.32 0.09 0.078 0.49 0.07
15.011 1.31 0.09 0.083 0.52 0.06
20.011 1.43 0.08 0.098 0.67 0.04
25.011 1.66 0.05 0.107 0.84 0.02
30.011 1.54 0.06 0.102 0.75 0.03
35.011 1.47 0.07 0.085 0.60 0.05
40.011 1.10 0.14 0.067 0.35 0.10
45.011 1.30 0.10 0.074 0.46 0.07
50.011 1.41 0.08 0.079 0.53 0.06
55.011 1.37 0.09 0.081 0.53 0.06
60.011 1.57 0.06 0.087 0.65 0.04
65.011 2.26 0.012 0.136 1.44 0.005
70.011 3.05 0.0011 0.178 2.55 0.0003
75.011 3.24 0.0006 0.192 2.92 0.00012
80.011 2.34 0.010 0.138 1.53 0.004
85.011 1.74 0.04 0.099 0.82 0.03
90.011 1.45 0.07 0.084 0.58 0.05
95.011 0.88 0.2 0.060 0.25 0.14
There is now a single linkage peak around 75cM (LOD of 2.92). The original peak around
20cM has completely disappeared, and was simply an artifact of linkage disequilibrium
between markers. Thus, there is some good evidence for a single linkage peak in these
data (at around 75cM). The analysis ignoring linkage disequilibrium, which showed an
additional peak at around 20cM was quite inaccurate.
If you want to model linkage disequilibrium, but do not have a file describing
preset clusters for your SNP mapping panel, MERLIN provides two options for automatically
clustering markers. The --distance k option inserts a cluster breakpoint
between markers that are less than k cM apart (that is, all consecutive markers
spaced less than k cM are placed into a cluster). The --rsq treshold option
calculates pairwise r2 between neighboring markers and creates
a cluster joining markers for which pairwise r2 > threshold and
all intervening markers.
To explore these alternative options, try the following command lines:
prompt> merlin -d snp-scan.dat -p snp-scan.ped -m snp-scan.map --npl --grid 5 --rsq 0.1 --cfreq
prompt> merlin -d snp-scan.dat -p snp-scan.ped -m snp-scan.map --npl --grid 5 --dist 3 --cfreq
prompt> merlin -d snp-scan.dat -p snp-scan.ped -m snp-scan.map --npl --grid 5 --clusters snp-scan.clusters-only --cfreq
The first command-line, will search for markers for which r2 is > 0.10 and define clusters
including each identified pair and the intervening markers. The second command-line will group
markers that are less than 3 cM apart into a cluster. The final command-line will use
the cluster definitions in the snp-scan.clusters-only file, but estimate haplotype frequencies
from the available genotype data. In each case, the --cfreq flag requests that the estimated
clusters and their frequencies should be saved to a file.
That is it! You should be on your way to modeling linkage disequilibrium between markers
in your own data, so as to make the best use of available SNP mapping panels.
To learn about other analyses options, you might want to check the non-parametric
linkage analysis or parametric linkage analysis sections, or proceed
haplotyping, simulation or ibd
estimation sections.
| 