|
1. Introduction
vcfCodingSnps is a variant annotation tool that annotates genetic variants such as single nucleotide polymorphisms (SNPs) and short insersion and deletions (INDELs) in a VCF format input file. It takes a VCF as an input and generates an annotated VCF file as an output.
Given a VCF SNP (or short INDEL) file, vcfCodingSnps will annotate each single varaint in a row according to a user specified gene list and a reference genome. The gene list and the reference genome that user provided can be of various gene tracks and assemblies. The latest version takes gene list tracks such as UCSC known genes, RefSeq genes, Genecode genes, CCDS genes and Emsembl genes, and the assembly of the gene list and the reference genome can be of either hg16, hg17, hg18 or hg19. One can explore UCSC genome browser for a better understanding of different tracks and assemblies. By default vcfColdingSnps uses a hg18 UCSC known gene list and the hg18 reference genome. It also provides versions of other tracks and assemblies at the user's conveinience so that they don't need to download those themselves. One can find more detailed information about input files here.
For a single variant in the input VCF file, vcfCodingSnps annotates it within each gene region (including upstream and downstream regions of the gene) that covers the variant and outputs it into the output VCF file; while for each single gene in the input gene list, vcfCodingSnps lists the annotated results for the variants that lie in that gene region in the output log file to fersilitate gene based analyses. More information about output files are provided here.
2. Functional Categories
The functional categories of annotation that integrated in vcfCodingSnps in the current version includes:
* For single nucleotide polymorphisms (SNPs) :
Category |
Definition
used in vcfCoding Snps |
| stop gained |
a SNP in coding sequence and introducing a
TAG, TAA, or TGA stop codon |
| stop lost |
a SNP in coding sequence and causing a loss of a
TAG, TAA, or TGA stop codon |
| non-synonymous
coding |
a SNP in coding sequence, located in a codon
resulting in a change of amino acid, excluding
SNPs that can be defined as either stop gained or stop lost |
| synonymous coding |
a SNP in coding sequence, located in a codon
that not resulting in a change of amino acid |
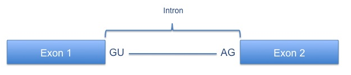
| essential splice site |
a SNP changing the
highly conserved GU in the first two basepairs of the intron or (AG) in the last
two basepair of the intron
 |
| splice site |
a SNP occurring in 3 - N1 basepairs into the intron, or N2 basepairs into the exon . N1 by default is 8, N2 by default is 3. N1 and N2 can be defined by user through option --n1 --n2. |
| 5' UTR |
a SNP located within the
5' UTR of a transcript |
| 3' UTR |
a SNP located within the
3' UTR of a transcript |
| intronic |
a SNP in the intron of a
known gene, and cannot be defined as essential splice site or splice site |
| upstram |
A SNP located within N kb from the transcript start site (5'-end) of a known gene, N by default is 5 and can be defined by user through option --ns |
| downstream |
A SNP located within N kb from the transcript end site (3'-end) of a known gene, N by default is 5 and can be defined by user through option --ns |
| introgenic |
A SNP not located within a known gene and also not identified as upstream or downstream of a knowngene |
A toy example is provided here for illustrating how SNPs are annotated into above categories. Figure1 is a fregment of reference genome with a gene located from bp 2 to 67 (from "Gene start" to "Gene end"). The upper case letter in each box is the reference allele at that bp position on the reference genome. The blocks in blue color represent exons in the gene. The yellow and orange bars below the box array represent condons in the coding region. We annotate ten SNPs marked by bold red arrows above correponding bp positions. The alternative alleles of each SNP and the annotation results are given in Table 1 below.
Figure 1. A toy example of annotating 10 SNPs in a gene on the reference genome
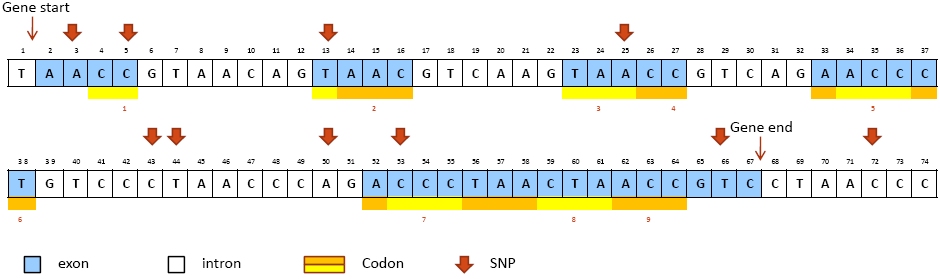 |
Table 1. Alternative alleles and annotation results
| Pos |
Alt SNP |
Ref SNP |
Alt SNP Codon |
Ref SNP Codon |
Alt SNP AA |
Ref SNP AA |
Anno Type |
| 3 |
G |
A |
-- |
-- |
-- |
-- |
5'UTR |
| 5 |
A |
C |
CAT |
CCT |
His |
Pro |
Non_Synonymous |
| 13 |
G |
T |
CCG |
CCT |
Pro |
Pro |
Synonymous |
| 25 |
C |
A |
TAC |
TAA |
Tyr |
Stop |
Stop Loss |
| 43 |
A |
C |
-- |
-- |
-- |
-- |
Splice Site |
| 44 |
G |
T |
-- |
-- |
-- |
-- |
Intronic |
| 50 |
C |
A |
-- |
-- |
-- |
-- |
Essential Splice Site |
| 53 |
A |
C |
ACC |
CCC |
Thr |
Pro |
Non_Synonymous |
| 66 |
C |
T |
-- |
-- |
-- |
-- |
3'UTR |
| 72 |
A |
C |
-- |
-- |
-- |
-- |
Downstram |
3. A Quick Start Quideline
Here is an example for the first time user of vcfCodingSnps. After installation, at the root folder of the package, type
./vcfCodingSnps.v1.5 -s example/example.input.vcf
and you would expect to see a screen output like:
##################################################################################################
vcfCodingSnps1.5 -- vcf SNP annotating tool
(c) 2010.5 Yanming Li, Goncalo Abecasis
Commend and (or) suggestions are welcome! Please send to liyanmin@umich.edu.
##################################################################################################
The following parameters are in effect:
Availabe Options
Input Files : --refgenome [referenceGenomes/genome.V36.fa],
--snpfile [example/example.input.vcf],
--genefile [geneLists/UCSCknownGene.B36.txt], --n1 [8],
--n2 [3], --ns [5]
Output Files : --outfile [vcfCodingSNP.out.vcf], --log [ON]
Reading chromosome >1 dna:chromosome chromosome:NCBI36:1:1:247249719:1...
Reading chromosome >2 dna:chromosome chromosome:NCBI36:2:1:242951149:1...
Reading chromosome >3 dna:chromosome chromosome:NCBI36:3:1:199501827:1...
Reading chromosome >4 dna:chromosome chromosome:NCBI36:4:1:191273063:1...
Reading chromosome >5 dna:chromosome chromosome:NCBI36:5:1:180857866:1...
Reading chromosome >6 dna:chromosome chromosome:NCBI36:6:1:170899992:1...
Reading chromosome >7 dna:chromosome chromosome:NCBI36:7:1:158821424:1...
Reading chromosome >8 dna:chromosome chromosome:NCBI36:8:1:146274826:1...
Reading chromosome >9 dna:chromosome chromosome:NCBI36:9:1:140273252:1...
Reading chromosome >10 dna:chromosome chromosome:NCBI36:10:1:135374737:1...
Reading chromosome >11 dna:chromosome chromosome:NCBI36:11:1:134452384:1...
Reading chromosome >12 dna:chromosome chromosome:NCBI36:12:1:132349534:1...
Reading chromosome >13 dna:chromosome chromosome:NCBI36:13:1:114142980:1...
Reading chromosome >14 dna:chromosome chromosome:NCBI36:14:1:106368585:1...
Reading chromosome >15 dna:chromosome chromosome:NCBI36:15:1:100338915:1...
Reading chromosome >16 dna:chromosome chromosome:NCBI36:16:1:88827254:1...
Reading chromosome >17 dna:chromosome chromosome:NCBI36:17:1:78774742:1...
Reading chromosome >18 dna:chromosome chromosome:NCBI36:18:1:76117153:1...
Reading chromosome >19 dna:chromosome chromosome:NCBI36:19:1:63811651:1...
Reading chromosome >20 dna:chromosome chromosome:NCBI36:20:1:62435964:1...
Reading chromosome >21 dna:chromosome chromosome:NCBI36:21:1:46944323:1...
Reading chromosome >22 dna:chromosome chromosome:NCBI36:22:1:49691432:1...
Reading chromosome >23 dna:chromosome chromosome:NCBI36:X:1:154913754:1...
start mapping
mapping snp file... ...DONE! snp mapsize = 29642
mapping gene file... ...DONE! gene mapsize = 66803
start annotating... ...
DONE! Complete annotating!!!
|
Hope this could give you some flavor on how vcfCodingSnps works.
4. Command Line Options
| -s |
SNP file |
This option specifies the name of the input VCF-format SNP file |
| -r |
reference genome file |
This option specifies the name of the imput reference genome FASTA file. It should be of either NCBI release 36/hg18 or GRCH37/hg19 format. By default it will load NCBI36 reference genome. Users can chose to download other versions of reference genome files at the download page |
| -g |
gene file |
Specifies the name of the input gene file, by default use a gene file (UCSCknownGene.B36.txt) generated by UCSC genome browser |
| -o |
output file |
Specifies the name of the output VCF-format SNP file, by default will be named vcfCodingSNP.out.vcf |
| -l |
log file |
Specifies the name of the log file, log file gives more detailed information for each annotated SNP, by default will be named vcfCodingSNP.log |
| --n1 |
parameter |
user defined number of bps into intron for splice site, by default will be set to 8 |
| --n2 |
parameter |
user defined number of bps into extron for splice site, by default will be set to 3 |
| --ns |
parameter |
user defined number of kbps for the range of upstream or downstream of a gene, by default will be set t0 5 |
|