MERLIN Tutorial -- Association Analysis
Merlin can test for association between a SNP and one or more quantitative traits (if
you are interested in discrete traits, you should consider the
LAMP software package, which provides
discrete trait association
tests that integrate over missing genotypes in small pedigrees).
The association test implemented in Merlin includes an integrated genotype inference feature,
which can improve power when some genotypes are missing
(Burdick et al. 2006).
In this example, we will see how to carry out an association analysis using Merlin and how to
use the integrated genotype inference feature to estimate missing genotypes.
The association tests implemented in Merlin can be used to analyze genome-wide association scans,
or to study candidate regions. However, it is important to note that -- in contrast to standard
family-based association tests -- the test implemented in Merlin does not control for population
stratification. If population stratification is a concern, population membership should be included
as a covariate or genomic control methods should be used to adjust results.
Alright ... let's walk through the analysis of an exemplar dataset. The dataset consists of 107 individuals in 9 three generation pedigrees
(modelled after the CEPH pedigrees originally collected to help in linkage map
construction, and which were more recently used by the HapMap Consortium to build a haplotype map of the human genotype
and wre also used to study the genetics of gene expression by multiple independent groups).
The data consist of genotypes for 20 SNP markers, with an average heterozigosity of about 40%. Six of the markers are genotyped
for all individuals, the remaining 14 are genotyped in only 50-54 individuals. The dataset is organized
into 3 files, a data file (assoc.dat), a pedigree file (assoc.ped), and a map file (assoc.map). All
of these are available in the examples subdirectory of the Merlin distribution and, as usual, you can check their contents
using pedstats.
To run Merlin for the association analysis, we need to specify the usual set of data (-d parameter), a pedigree (-p parameter), and a map files
(-m parameter). In addition, we need to request one of the following association tests: a score test (--fastAssoc) or a likelihood-ratio test
(--assoc). The score test (--fastAssoc) is rapid and ideal for screening very large numbers of markers (for example, in a first pass analysis of a genome-wide
association (GWA) scan), whereas the more accurate likelihood-ratio test (--assoc) can be used to evaluate smaller numbers of markers (for example, in candidate regions
selected for
follow up analyses). In datasets that include only small pedigrees or when the effects being evaluated are small, the two tests will give very similar results.
In this example, we will first try the --fastAssoc option, using the following command line:
prompt> merlin -d assoc.dat -p assoc.ped -m assoc.map --fastAssoc
After running the command, you should first see a summary of the currently selected and available
options. At the end of your output, you should see the following table of results:
Phenotype: mRNA [FAST-ASSOC] (9 families, h2 = 15.99%)
==============================================================================
Position Marker Allele Effect H2 LOD pvalue
56.077 SNP1 3 0.168 5.93% 1.186 0.02
56.081 SNP2 2 0.168 5.71% 1.185 0.02
56.081 SNP3 2 0.048 0.26% 0.051 0.6
56.499 SNP4 1 -0.207 4.39% 0.906 0.04
56.501 SNP5 3 0.172 4.11% 0.795 0.06
56.509 SNP6 4 -0.058 0.64% 0.129 0.4
56.938 SNP7 3 0.026 0.16% 0.032 0.7
56.941 SNP8 4 0.026 0.17% 0.026 0.7
56.949 SNP9 3 0.002 0.00% 0.000 1.0
57.114 SNP10 1 -0.122 1.51% 0.205 0.3
57.118 SNP11 3 0.497 47.02% 8.522 3.7e-10
57.123 SNP12 2 0.315 23.60% 4.343 7.7e-06
57.126 SNP13 4 0.315 23.60% 4.343 7.7e-06
57.590 SNP14 2 0.115 3.14% 0.633 0.09
57.600 SNP15 3 0.088 1.82% 0.312 0.2
57.610 SNP16 3 0.088 1.82% 0.312 0.2
59.410 SNP17 1 0.092 1.65% 0.344 0.2
59.417 SNP18 4 -0.026 0.15% 0.027 0.7
59.418 SNP19 1 0.166 4.69% 0.750 0.06
59.784 SNP20 3 0.178 7.52% 1.432 0.010
Peak --> SNP11 3 0.497 47.02% 8.522 3.7e-10
The table summarizes the --fastAssoc analysis of phenotype "mRNA". The 7 columns are the position and name of the SNP being tested (markers
with more than two alleles will be skipped), the allele being tested,
the estimated effect of the allele, the proportion of total variance explained by the SNP, a LOD score statistic summarizing evidence for association
and its corresponding p-value.
The last row highlights the strongest association among all SNPs examined. In this case, it looks like every copy of allele '3' at SNP11 increases
phenotypic values by approximately 0.5 units. Overall, the SNP explains 47% of the variation in mRNA levels for the trait and is associated with a
LOD score of about 8.5. Examining the detailed output, you'll see that two nearby SNPs are also strongly associated -- these are likely in linkage
disequilibrium with the SNP that shows strongest association.
Since the results look interesting, it seems worthwhile to follow-up the score test with a more time-consuming
maximum likelihood analysis. In large datasets, you could focus this follow-up analysis on the most promising SNPs using
the --start and --stop options. If you do that, all SNPs outside the region specified by -start and --stop
will still be used for inference of missing genotypes, but they
won't be tested for association. In this case, there are only 20 SNPs to analyse and the maximum likleihood analysis shouldn't be
too time consuming. Since we are dealing with relative large pedigrees (each pedigree has an average of >10 individuals) and a relatively large
effect (the SNP explains nearly half of the variation in phenotypic values), we expect that the maximum likleihood analysis will provide
us with more accurate results. To carry out the follow-up analysis, try the following command
line:
prompt> merlin -d assoc.dat -p assoc.ped -m assoc.map --assoc
You should see the following results table towards the end of Merlin output:
Phenotype: mRNA [ASSOC] (9 families, h2 = 15.99%)
===============================================================================
---- LINKAGE TEST RESULTS ---- ----------- ASSOCIATION TEST RESULTS ----------
Position H2 LOD pvalue Marker Allele Effect H2 LOD pvalue
56.077 44.1% 2.12 0.0009 SNP1 3 0.182 6.74% 1.06 0.03
56.081 44.2% 2.12 0.0009 SNP2 2 0.182 6.50% 1.07 0.03
56.081 44.2% 2.12 0.0009 SNP3 2 0.058 0.37% 0.05 0.6
56.499 46.5% 2.56 0.0003 SNP4 1 -0.192 3.65% 0.67 0.08
56.501 46.5% 2.56 0.0003 SNP5 3 0.178 4.25% 0.58 0.10
56.509 46.5% 2.57 0.0003 SNP6 4 -0.031 0.18% 0.02 0.7
56.938 46.8% 2.87 0.00014 SNP7 3 0.053 0.67% 0.10 0.5
56.941 46.8% 2.87 0.00014 SNP8 4 0.061 0.87% 0.10 0.5
56.949 46.8% 2.87 0.00014 SNP9 3 0.020 0.08% 0.01 0.8
57.114 46.8% 2.87 0.00014 SNP10 1 -0.114 1.28% 0.14 0.4
57.118 46.8% 2.87 0.00014 SNP11 3 0.477 42.46% 8.48 4.1e-10
57.123 46.8% 2.87 0.00014 SNP12 2 0.283 18.66% 2.74 0.0004
57.126 46.8% 2.87 0.00014 SNP13 4 0.283 18.65% 2.74 0.0004
57.590 46.8% 2.87 0.00014 SNP14 2 0.098 2.24% 0.34 0.2
57.600 46.8% 2.87 0.00014 SNP15 3 0.066 1.01% 0.13 0.4
57.610 46.8% 2.87 0.00014 SNP16 3 0.066 1.01% 0.13 0.4
59.410 47.0% 2.87 0.00014 SNP17 1 0.094 1.69% 0.26 0.3
59.417 47.0% 2.87 0.00014 SNP18 4 -0.042 0.39% 0.05 0.6
59.418 47.0% 2.87 0.00014 SNP19 1 0.153 3.92% 0.48 0.14
59.784 47.1% 2.87 0.00014 SNP20 3 0.158 5.86% 0.88 0.04
Peak --> SNP11 3 0.477 42.46% 8.48 4.1e-10
The two commands we just walked through, --assoc and --fastAssoc, are the two you will use most often when testing for association.
The commands work within Merlin for autosomal analysis, and also within Minx for the analysis of X-linked markers. You will often find it useful to
combine them with the --pdf option (which generates a graphical summary of their results) and the --inverseNormal option (which automatically transforms
traits so they follow a smooth normal distribution).
Below, we describe how to carry out sequential association analyses (to identify multiple SNPs that are associated with the trait of interest) and how
to get Merlin to output imputed genotype distirbutions for analysis in other programs. You may decide to only read about those options later, after you have
tried out the --assoc and -fastassoc options on your own data.
Int his relatively small dataset, it was convenient to browse results in Merlin's screen output. When analyzing very large datasets, you may find
the --tabulate option more convenient. This generates a tab-delimited output file that can be readily imported into many downstream analysis programs.
Advanced Exercise - Sequential Association Analysis
Merlin usually tests for association one SNP at a time. After identifying the most strongly associated SNP, it is often interesting to check whether
this SNP can account for the association at other neighboring SNPs and to search for other independently associated SNPs. One way to do this is to
gradually refine our trait model. We might start with a model that includes only environmental covariates and search for the best associated SNP. After this
SNP is identified, we might add it to the list of covariates and re-evaluate the evidence for association at all other SNPs. And so on ...
To customize the covariate list for qunatitative trait association analysis, we use the --custom option. This option specifies a file that describes
a series of customized trait models. Each model starts with a trait name (indicated by the TRAIT keyword) and is optionally followed by a list of covariates
(indicated by the COVARIATES keyword). To carry out a sequential association analysis, we start with a very simple custom model file (in this example, we will
use the assoc.tbl file) and gradually refine it by including the best SNP from each round as a covariate.
To get things started, run the command:
prompt> merlin -d assoc.dat -p assoc.ped -m assoc.map --custom assoc.tbl --assoc
The assoc.tbl file is very simple, and includes a single line of interest:
< Contents of assoc.tbl file >
TRAIT mRNA
< End of assoc.tbl file >
Thus, it simply specifies that Merlin should run a simple analysis for the trait mRNA
wiht no covariates. When you run Merlin you should see the following output:
CUSTOM QUANTITATIVE TRAIT MODEL #1
===================================
TRAIT: mRNA
No covariates
Phenotype: mRNA [ASSOC] (9 families, h2 = 15.99%)
===============================================================================
---- LINKAGE TEST RESULTS ---- ----------- ASSOCIATION TEST RESULTS ----------
Position H2 LOD pvalue Marker Allele Effect H2 LOD pvalue
56.077 44.1% 2.12 0.0009 SNP1 3 0.182 6.74% 1.06 0.03
56.081 44.2% 2.12 0.0009 SNP2 2 0.182 6.50% 1.07 0.03
56.081 44.2% 2.12 0.0009 SNP3 2 0.058 0.37% 0.05 0.6
56.499 46.5% 2.56 0.0003 SNP4 1 -0.192 3.65% 0.67 0.08
56.501 46.5% 2.56 0.0003 SNP5 3 0.178 4.25% 0.58 0.10
56.509 46.5% 2.57 0.0003 SNP6 4 -0.031 0.18% 0.02 0.7
56.938 46.8% 2.87 0.00014 SNP7 3 0.053 0.67% 0.10 0.5
56.941 46.8% 2.87 0.00014 SNP8 4 0.061 0.87% 0.10 0.5
56.949 46.8% 2.87 0.00014 SNP9 3 0.020 0.08% 0.01 0.8
57.114 46.8% 2.87 0.00014 SNP10 1 -0.114 1.28% 0.14 0.4
57.118 46.8% 2.87 0.00014 SNP11 3 0.477 42.46% 8.48 4.1e-10
57.123 46.8% 2.87 0.00014 SNP12 2 0.283 18.66% 2.74 0.0004
57.126 46.8% 2.87 0.00014 SNP13 4 0.283 18.65% 2.74 0.0004
57.590 46.8% 2.87 0.00014 SNP14 2 0.098 2.24% 0.34 0.2
57.600 46.8% 2.87 0.00014 SNP15 3 0.066 1.01% 0.13 0.4
57.610 46.8% 2.87 0.00014 SNP16 3 0.066 1.01% 0.13 0.4
59.410 47.0% 2.87 0.00014 SNP17 1 0.094 1.69% 0.26 0.3
59.417 47.0% 2.87 0.00014 SNP18 4 -0.042 0.39% 0.05 0.6
59.418 47.0% 2.87 0.00014 SNP19 1 0.153 3.92% 0.48 0.14
59.784 47.1% 2.87 0.00014 SNP20 3 0.158 5.86% 0.88 0.04
Peak --> SNP11 3 0.477 42.46% 8.48 4.1e-10
Refined association models stored in [merlin-assoc-covars.*]
The results should be identical to the ones from the earlier analysis, used to demonstrate
the --assoc option. However, the key thing for us is the final line of output -- which indicates
Merlin has automatically generated a set of files that will help in our sequential analysis. The set
inlcudes three files. One of these, merlin-assoc-covars.tbl, includes a refined trait model that now includes SNP11
as a covariate. The other two, merlin-assoc-covars.dat and merlin-assoc-covars.ped, include an appropriately
coded covariate which indicates the number of copies of allele '3' at SNP11 carried by each individual.
To continue our sequential analysis, we first merge the covariate into the original pedigree file and rename
merlin-assoc-covars.tbl so that it is not overwritten when we next run Merlin. Run the following series of commands:
prompt> pedmerge assoc merlin-assoc-covars assoc-stage2
prompt> mv merlin-assoc-covars.tbl assoc-stage2.tbl
The first command combines the original pedigree file with the covariate data automatically generated by Merlin. The
second command renames the trait model file generated by Merlin, so it is not overwritten on our next analysis (on windows, you should
replaced the mv command with the move command). We are now ready to run the second round association analysis:
prompt> merlin -d assoc-stage2.dat -p assoc-stage2.ped -m assoc.map --custom assoc-stage2.tbl --assoc
The results of this second round (pasted below), show that SNPs 12 and 13 (which showed some evidence for association in the first pass analysis)
are no longer significantly associated -- their effects were likely a consequence of their closeness to SNP11. The only SNP that shows marginal
evidence for association is SNP20, but this is likely not significant after adjusting for multiple testing. Thus, we stop our sequential analysis here!
CUSTOM QUANTITATIVE TRAIT MODEL #1
===================================
TRAIT: mRNA
COVARIATES: SNP11
Phenotype: mRNA [ASSOC] (9 families, h2 = 0.00%)
===============================================================================
---- LINKAGE TEST RESULTS ---- ----------- ASSOCIATION TEST RESULTS ----------
Position H2 LOD pvalue Marker Allele Effect H2 LOD pvalue
56.077 0.0% 0.00 0.5 SNP1 3 0.037 0.49% 0.10 0.5
56.081 0.0% 0.00 0.5 SNP2 2 0.037 0.47% 0.10 0.5
56.081 0.0% 0.00 0.5 SNP3 2 0.027 0.14% 0.03 0.7
56.499 0.0% 0.00 0.5 SNP4 1 -0.103 1.78% 0.38 0.2
56.501 0.0% 0.00 0.5 SNP5 3 -0.022 0.11% 0.02 0.8
56.509 0.0% 0.00 0.5 SNP6 4 -0.089 2.46% 0.62 0.09
56.938 0.0% 0.00 0.5 SNP7 3 -0.009 0.03% 0.01 0.9
56.941 0.0% 0.00 0.5 SNP8 4 -0.009 0.03% 0.01 0.9
56.949 0.0% 0.00 0.5 SNP9 3 0.037 0.45% 0.08 0.5
57.114 0.0% 0.00 0.5 SNP10 1 -0.090 1.34% 0.21 0.3
57.118 0.0% 0.00 0.5 SNP11 3 - - - -
57.123 0.0% 0.00 0.5 SNP12 2 -0.030 0.35% 0.04 0.7
57.126 0.0% 0.00 0.5 SNP13 4 -0.030 0.35% 0.04 0.7
57.590 0.0% 0.00 0.5 SNP14 2 0.101 3.98% 0.91 0.04
57.600 0.0% 0.00 0.5 SNP15 3 0.091 3.20% 0.63 0.09
57.610 0.0% 0.00 0.5 SNP16 3 0.091 3.20% 0.63 0.09
59.410 0.0% 0.00 0.5 SNP17 1 -0.001 0.00% 0.00 1.0
59.417 0.0% 0.00 0.5 SNP18 4 0.013 0.06% 0.01 0.8
59.418 0.0% 0.00 0.5 SNP19 1 0.091 2.34% 0.41 0.2
59.784 0.0% 0.00 0.5 SNP20 3 0.119 5.58% 1.13 0.02
Peak --> SNP20 3 0.119 5.58% 1.13 0.02
Refined association models stored in [merlin-assoc-covars.*]
Advanced Exercise - Standalone Genotype Inference
In this more detailed analysis, Merlin first evaluates the evidence for linkage at each position. The results are summarized in the
first 4 columns of the summary table, which show the position of the SNP being tested, the proportion of variance that is explained by IBD sharing
at that position in a variance component linkage analysis and the corresponding LOD score and p-value (the --vc option provides more
detailed linkage analysis results). Because we are examining a relatively small region, you will notice that the linkage signal changes only very
gradually and is nearly flat for most of the region. The next set of columns summarizes results of the association test. You will see the name of the SNP and allele being tested,
the estimated effect of the allele, the proportion of the trait variance it explains, and
finally the LOD score and p-value evaluating the evidence for association. In this case, the
--assoc option found even stronger evidence for association (as expected, since the SNP being tested is very close
to the gene encoding the mRNA levels we measured).
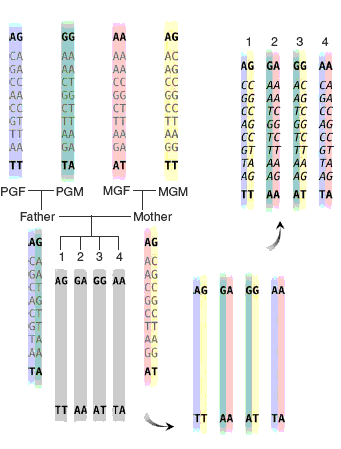
One unique feature of association tests in MERLIN is that missing genotypes are imputed and incorporated in the
association test. A cartoon illustration of the procedure is provided in the figure above (adapted from
Burdick et al. 2006).
In the figure, all missing genotypes can be inferred by using information from flanking markers.
Even when this is not the case, missing genotypes can often be estimated using information at other
markers and family relationships. In principle,
genotype inference can substantially improve power of association tests. In fact, we expect that
when a genome-wide association scan follows
a linkage study, only a porportion of individuals may need to be genotyped and genotypes of the remaining family members
can be estimated computationally.
Merlin allows genotype inference to be decoupled from association tests.
To estimate missing genotypes, use the --infer parameter. Details
of estimated genotypes will be stored in a pair of files, merlin-infer.dat and
merlin-infer.ped. Try the following command line:
prompt> merlin -d assoc.dat -p assoc.ped -m assoc.map --infer
In the inferred pedigree file (saved as merlin-infer.ped here), each genotype is described in 5 columns: the most likely genotype,
the expected number of copies for the tested allele (0, 1, or 2 if the genotype is observed; but fractional counts may occur for missing
genotyeps), and the posterior probabilities for
the three alternative genotypes. This information can be useful to other programs that can use imputed genotype distributions to test for
association, but that are not themselves equipped with an integrated genotype inference feature.
This tutorial was written by Weimin Chen and Goncalo Abecasis. Hope you enjoyed it!
| 