MERLIN Tutorial -- Error detection
Genotyping errors can lead to misleading inferences about gene
flow in pedigrees and greatly reduce the effectiveness of pedigree
analysis. In this section, we will use MERLIN to conduct a sensitivity
analysis of the likelihood and identify problem genotypes.
You can find the simulated data set for this section
in the examples subdirectory of the MERLIN distribution or in the
download page.
The dataset consists of a simulated 5-cM scan of chromosome 24 in
200 affected sib-pair families and is organized into 3 files, a
data file (error.dat), a pedigree file (error.ped) and
a map file (error.map). An overview of MERLIN input files is available
elsewhere.
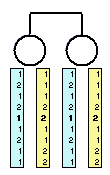
How does error detection work?
Before conducting the error detection analysis, we will review the
basic principles behind it. Consider the simple pedigree to the left,
with two siblings genotyped at several markers. Since their genotypes
are identical at all markers, it seems quite likely that they share
the stretch of chromosome under investigation.
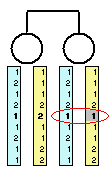
Now, consider what happens if we change the genotype for a single
marker (indicated by the red circle)... This marker now contradicts
information provided by all others, indicating that perhaps one of
the parents carried two nearly identical copies of the chromosome or two
recombination events occurred.
In the first example, inference about inheritance is relatively
consistent at all markers, while in the second example inference about
inheritance is strongly influenced by the single genotype. Intuitively,
the first outcome seems much more plausible.
MERLIN finds genotypes that provide information about gene flow
in a pedigree that contradicts information provided by other available
data. MERLIN considers all available data simultaneously (not just
pairs of individuals) so that error detection improves in accuracy
in larger pedigrees. Genotypes flagged by MERLIN are likely to be
errors and are certainly worth checking!
Error detection using MERLIN
To run error detection using merlin, we need to provide an input
pedigree file (-p command line option) and matching data and
map files (-d and -m options) and request an error detection
analysis (--error option):
prompt> merlin -d error.dat -p error.ped -m error.map --error
Try it out! You should see the merlin banner and a summary of selected
options, followed by a list of unlikely genotypes. In this case, this
is the list:
Family: 2 - Founders: 2 - Descendants: 2 - Bits: 2
MRK11 genotype for individual 3 is unlikely [0.003848]
MRK11 genotype for individual 4 is unlikely [0.003848]
Family: 73 - Founders: 2 - Descendants: 2 - Bits: 2
MRK17 genotype for individual 3 is unlikely [0.008866]
MRK17 genotype for individual 4 is unlikely [0.008866]
Family: 81 - Founders: 2 - Descendants: 2 - Bits: 2
MRK8 genotype for individual 3 is unlikely [0.001567]
MRK8 genotype for individual 4 is unlikely [0.001567]
Family: 94 - Founders: 2 - Descendants: 2 - Bits: 2
MRK12 genotype for individual 3 is unlikely [0.002101]
MRK12 genotype for individual 4 is unlikely [0.002101]
Family: 136 - Founders: 2 - Descendants: 2 - Bits: 2
MRK16 genotype for individual 3 is unlikely [0.008330]
MRK16 genotype for individual 4 is unlikely [0.008330]
Family: 162 - Founders: 2 - Descendants: 2 - Bits: 2
MRK14 genotype for individual 3 is unlikely [0.003037]
MRK14 genotype for individual 4 is unlikely [0.003037]
Family: 164 - Founders: 2 - Descendants: 2 - Bits: 2
MRK6 genotype for individual 3 is unlikely [0.001805]
MRK6 genotype for individual 4 is unlikely [0.001805]
Unlikely genotypes listed in file [merlin.err]
In this data set with 20 markers and 200 sib-pair families, MERLIN flagged
7 pairs of unlikely genotypes. Since we are dealing with sib-pairs, errors are not
pinpointed to specific individuals (all that we can tell is that at least one of
the siblings is likely to have an erroneous genotype in each family!).
In a real-life setting it would be worthwhile re-checking genotype
assays for these individuals. In this case, we will simply run pedwipe
to erase genotypes that are flagged as problematic. Run:
prompt> pedwipe -d error.dat -p error.ped
Pedwipe retrieves a list of unlikely genotypes from the merlin.err file and
removes them from the data. A new set of data and pedigree files is created, named
wiped.dat and wiped.ped. You can get a feel for the impact
of these 7 problematic genotypes on linkage analysis by running a non-parametric
linkage analysis before and after their removal:
prompt> merlin -d error.dat -p error.ped -m error.map --npl
(...excerpt of results before removing problematic genotypes...)
Phenotype: affection [ALL] (200 families)
============================================================
Pos Zmean pvalue delta LOD pvalue
42.144 2.16 0.02 0.186 1.24 0.008
47.412 2.39 0.008 0.204 1.51 0.004
52.680 2.57 0.005 0.214 1.69 0.003
57.948 1.72 0.04 0.145 0.76 0.03
63.216 1.19 0.12 0.106 0.39 0.09
prompt> merlin -d wiped.dat -p wiped.ped -m error.map --npl
(...excerpt of results after removing problematic genotypes...)
Phenotype: affection [ALL] (200 families)
============================================================
Pos Zmean pvalue delta LOD pvalue
42.144 2.24 0.012 0.191 1.32 0.007
47.412 2.48 0.007 0.209 1.60 0.003
52.680 2.87 0.002 0.237 2.10 0.0009
57.948 2.10 0.02 0.175 1.13 0.011
63.216 1.47 0.07 0.127 0.57 0.05
The seven problematic genotypes (out of 8,000 total genotypes),
cause a 0.4 change in the Kong and Cox allele sharing LOD score!
To learn about estimating false positive rates for error detection and
linkage analysis you should proceed to the simulation section.
Alternatively, you may want to learn more about linkage
analysis, haplotyping or
ibd estimation.
| 