MERLIN Tutorial -- Parametric Linkage Analysis
Linkage analysis tests for co-segregation of a chromosomal region
and a trait locus of interest. In parametric linkage analysis, a specific
disease model is used to describe segregation of the trait locus.
In this section, we will walk through a parametric linkage analysis using MERLIN.
For this example, we will use a simulated data set that you will find
in the examples subdirectory of the MERLIN distribution or in the
download page.
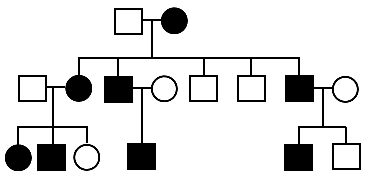
The dataset consists of a 10-cM scan of candidate chromosome
in a single pedigree
where a rare dominant disorder is segregating (the pedigree is picture above).
Ten microsatellite markers,
each with 4 equally frequent alleles, were genotyped in all pedigree
members. The genotypes and phenotypes are described in 3 files, a
data file (parametric.dat), a pedigree file (parametric.ped)
and a map file (parametric.map). An overview of MERLIN input files
is available elsewhere.
The recommended first step in any analysis is to verify that input
files are being interpreted correctly. So let's start by running
pedstats... Pedstats requires an input data file (-d parameter) and
pedigree file (-p parameter):
prompt> pedstats -d parametric.dat -p parametric.ped
By examining the abbreviated pedstats output below, you should be
able to confirm that there is a single pedigree, with a total of
16 individuals (8 of these individuals are affected), and that there
is no missing phenotype or genotype data.
Pedigree Statistics - 0.5.4
(c) 1999-2005 Goncalo Abecasis, 2002-2005 Jan Wigginton
The following parameters are in effect:
Pedigree File : parametric.ped (-pname)
Data File : parametric.dat (-dname)
PEDIGREE STRUCTURE
==================
Individuals: 16
Founders: 5 founders, 11 nonfounders
Gender: 6 females, 10 males
Families: 1
Generations
Average: 3.00 (3 to 3)
Distribution: 3 (100.0%), 0 (0.0%) and 1 (0.0%)
AFFECTION STATISTICS
=====================
[Diagnostics] [Founders] Prevalence
VERY_RARE_DISEASE 16 100.0% 5 100.0% 50.0%
Total 16 100.0% 5 100.0%
MARKER GENOTYPE STATISTICS
===========================
[Genotypes] [Founders] Hetero
MRK1 16 100.0% 5 100.0% 87.5%
MRK2 16 100.0% 5 100.0% 75.0%
(...statistics for other markers would appear here...)
MRK10 16 100.0% 5 100.0% 75.0%
Total 160 100.0% 50 100.0% 78.1%
The pedigree and data file seem to be okay. In addition to the standard
Merlin input files, parametric linkage analyses
require disease locus parameters to be specified in a separate text file. This text
file has one row for each of the disease models to be evaluated, and
can include as many different models as available memory allows. For this analysis,
the file parametric.model specifies a single rare dominant disease model.
Here are its contents:
| Affection |
Disease Allele Frequency |
Penetrances |
Model Name |
| VERY_RARE_DISEASE |
0.0001 |
0.0001,1.0,1.0 |
Rare_Dominant |
In general, the file should be tab or space delimited, with 4 fields: affection
status label (matching the data file), disease allele frequency, probability of
being affected for individuals with 0, 1 and 2 copies of the disease allele (penetrances),
and finally a label for the analysis model. A header line is included in the
table above, for readability, but is not required. This file can also specify
penetrance functions that depend on a covariate,
such as age.
Okay ... let's run merlin! We will need to specify an input data file
(-d parameter), pedigree file (-p parameter) and map file
(-m parameter) as well as the file with trait model parameters
(--model command line option). Since parametric linkage LOD scores
tend to dip at marker locations, we will request an analyses at
three equally spaced locations between
each consecutive pair of markers with the --step 3 option. With all
these options, the command line will look like this:
prompt> merlin -d parametric.dat -p parametric.ped -m parametric.map --model parametric.model --step 3
After running the command, you should first see the MERLIN banner and a
summary of currently selected options:
MERLIN DEMO-VERSION - (c) 2000-2005 Goncalo Abecasis
The following parameters are in effect:
Data File : parametric.dat (-dname)
Pedigree File : parametric.ped (-pname)
Map File : parametric.map (-mname)
Allele Frequencies : ALL INDIVIDUALS (-f[a|e|f|file])
Data Analysis Options
General : --information, --likelihood, --model [parametric.model]
Positions : --steps [3], --maxStep, --minStep, --grid, --start,
--stop
Notice that allele frequencies were estimated by counting among
all individuals (the default). In this case, this does not matter
because all founders are genotyped. In practice, when analysing
small datasets such as this one, it might be a good
idea to genotype additional unrelated individuals to obtain
better estimates of allele frequencies or to use an
allele frequency file with custom frequencies.
After a minute or two, you should see analysis results at each
location:
Parametric Analysis, Model Dominant_Model
=======================================================
POSITION LOD ALPHA HLOD
(... some results edited to save space ...)
35.000 -1.291 0.000 0.000
37.500 2.037 1.000 2.037
40.000 2.263 1.000 2.263
42.500 2.358 1.000 2.358
45.000 2.388 1.000 2.388
47.500 2.201 1.000 2.201
50.000 1.959 1.000 1.959
52.500 1.585 1.000 1.585
55.000 -9.291 0.000 0.000
(... results continue at other locations...)
Each row indicates the estimated multipoint LOD score at a particular location.
This is followed by the estimate proportion of linked families (since there is
only one informative family in this sample, the proportion will always be 0.000
or 1.000), and the corresponding maximum heterogeneity LOD score. In this
case the maximum LOD score of 2.407 is observed at position 45.000, the position
of marker MRK5 in the map file.
Useful options for parametric linkage analyses options include requesting
output with marker names, instead of cM positions (--markerNames
option), requesting analysis along a grid of equally spaced locations (--grid
n for an n-cM grid) rather than at a fixed number of steps
between markers (--steps n for n-steps between consecutive markers),
or requesting a graph summarizing results (--pdf). Try them out! For example...
prompt> merlin -d parametric.dat -p parametric.ped -m parametric.map --model parametric.model --grid 1 --markerNames --pdf
... would calculate the parametric LOD scores for a 1-cM grid along the
chromosome and generate a PDF file with the resulting statistics.
That is it! That is all you need to get started with parametric linkage
analysis in Merlin. Remember to set your disease model carefully, as an appropriate
and careful choice of disease model is essential for parametric linkage
analyses.
To learn about other analyses options, you might want to check the non-parametric
linkage analysis section to find out how to conduct affecteds only linkage analyses. Or you could proceed
to the error detection (improves power!), haplotyping, simulation or ibd
estimation sections.
| 