MERLIN Input Files
MERLIN performs common pedigree analyses. Input files describe relationships between
individuals in your dataset, store marker genotypes, disease status and quantitative
traits and provide information on marker locations and allele frequencies.
MERLIN supports input files in either QTDT or LINKAGE format. Although the
two formats are very similar, in the discussion below we will focus on QTDT format.
Describing Relationships Between Individuals
Although pedigrees can become quite complex, all the information that is necessary to
reconstruct individual relationships in a pedigree file can be summarized in five items:
a family identifier, an individual identifier, a link to each parent (if available) and
finally an indicator of each individual's sex.
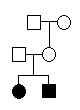 As an example of how family relationships are described, we will construct a pedigree
file for a small pedigree with two siblings, their parents and maternal grand-parents.
As an example of how family relationships are described, we will construct a pedigree
file for a small pedigree with two siblings, their parents and maternal grand-parents.
For this simple pedigree, the five key items take the following values:
FAMILY PERSON FATHER MOTHER SEX
example granpa unknown unknown m
example granny unknown unknown f
example father unknown unknown m
example mother granpa granny f
example sister father mother f
example brother father mother m
These key values constitute the first five columns of any pedigree
file. Because of restrictions in early genetic programs, text identifiers
are usually replaced by unique numeric values. After replacing each
identifier with unique integer and recoding sexes as 2 (female) and 1 (male),
this is what a basic space-delimited pedigree file would look like:
<contents of basic.ped>
1 1 0 0 1
1 2 0 0 2
1 3 0 0 1
1 4 1 2 2
1 5 3 4 2
1 6 3 4 1
<end of basic.ped>
A pedigree file can include multiple families. Each family can
have a unique structure, independent of other families in the dataset.
Describing Phenotypes and Genotypes
Usually the five standard columns are followed by various
types of genetic data, including phenotypes for discrete and quantitative
traits and marker genotypes.
Disease status is usually encoded in a single column as
U or 1 for unaffecteds,
A or 2 for affecteds, and
X or 0 for missing phenotypes.
Quantitative traits are encoded as numeric values with X
denoting missing values (it is also possible to use a peculiar numeric
value to flag missing phenotypes, but the procedure is prone to error
and not recommended).
Marker genotypes are encoded as two consecutive integers, one for each allele, optionally separated
by a "/", or since version 1.1 using the letters "A", "C", "T" and "G". To denote missing alleles,
either a 0, an X or an N can be used. The following are all valid genotype entries 1/1
(homozygote for allele 1), 0/0 (missing genotype), and 3 4 (heterozygote for alleles 3
and 4). In newer versions of Merlin A/A, A/C and C/C would also be valid
genotypes. For the X chromosome, males should be encoded as if they had two identical alleles.
This is what the previous pedigree file might look like after adding a
column for disease status, measurements for a quantitative trait and
genotypes for two markers:
<contents of basic2.ped>
1 1 0 0 1 1 x 3 3 x x
1 2 0 0 2 1 x 4 4 x x
1 3 0 0 1 1 x 1 2 x x
1 4 1 2 2 1 x 4 3 x x
1 5 3 4 2 2 1.234 1 3 2 2
1 6 3 4 1 2 4.321 2 4 2 2
<end of basic2.ped>
Notice that the two siblings (individuals 5 and 6 in the last two rows)
are marked as affected (value 2 in the sixth column), everyone else is marked
as unaffected (value 1 in the sixth column). The
quantitative trait (seventh column) takes values 1.234 and 4.321 for each sibling. Whereas
everyone is genotyped at the first marker, for the second marker, only
individuals 5 and 6 are genotyped.
Describing the pedigree file
Pedigree files can include any number of marker genotype, disease
status and quantitative trait variables, limited only by available
memory. Since each pedigree file has a unique structure (apart from
the first five columns), its contents must be described in a companion
data file.
The data file includes one row per data item in the pedigree file,
indicating the data type (encoded as M - marker, A - affection status,
T - Quantitative Trait and C - Covariate) and providing a one-word label
for each item. A data file for the pedigree above, which has one affection
status, followed by one quantitative trait and two marker genotypes might
read:
<contents of basic2.dat>
A some_disease
T some_trait
M some_marker
M another_marker
<end of basic2.dat>
You can get a summary description of any pair of pedigree and data files
using pedstats (included in the MERLIN distribution). To run pedstats
you must provide the name of your data file (-d command line option) and
pedigree file (-p command line option). In the MERLIN examples directory,
try the following command:
prompt> pedstats -d basic2.dat -p basic2.ped
TIP:In newer versions of Merlin and Pedstats, it is possible to combine multiple pedigree
and data files on the fly. This approach can be very convenient when analyzing multiple
different phenotypic subsets or when you want to separate genotypes by chromosome or by region. For
example, if your phenotypes are stored in files pheno.dat and pheno.ped and your genotypes are stored
in files geno.dat and geno.ped, you could combine them using the command line:
prompt> pedstats -d pheno.dat,geno.dat -p pheno.ped,geno.ped
Genetic Maps
To analyse genetic markers, MERLIN requires information on their
chromosomal location. This is usually provided in a map file.
If you are using sex-average maps, this file has one line per marker
with three columns, indicating chromosome, marker name and position
(in centiMorgans). If you are using sex-specific maps, you will
need two additional columns specifying the marker position along the female
and male genetic maps, respectively.
The data file and map file can include different sets of markers,
but markers that are absent from the map file will be ignored by
MERLIN. Here is what a typical map file looks like:
<contents of basic2.map>
CHROMOSOME MARKER POSITION
24 some_marker 123.4
24 another_marker 136.2
<end of basic2.map>
And here is a refined version of the map file including sex-specific
map positions for each marker:
<contents of file with sex-specific map>
CHROMOSOME MARKER POSITION FEMALE_POSITION MALE_POSITION
24 some_marker 123.4 146.8 100.0
24 another_marker 136.2 166.4 103.0
<end of sex-specific map>
Using separate data and map files makes for a very simple file structure and
allows MERLIN to analyse multiple chromosomes in a single run.
Allele Frequency Files
LINKAGE format data files specify the number of alleles at each locus
and their frequencies. When using QTDT format input files, MERLIN
estimates allele frequencies by counting alleles across all individuals.
If this is inappropriate for the analysis at hand you can request maximum
likelihood allele frequency estimates (-fm command line option),
specify equal allele frequencies (-fe), request estimates derived
by counting among founders only (-ff) or provide a custom allele
frequency file (-f filename option).
A custom allele frequency file indicates allele frequencies for all
marker alleles at each marker. For each marker, a single header line
naming the marker is followed by a list of allele frequencies, which
can take multiple lines.
Each header line is labelled M and includes the marker name. This header is
followed by a list of allele frequencies. There are two alternative formats for
lines in the allele frequency list:
- Classic format
- Lines in the allele frequency list are labelled F and list
frequencies for all alleles consecutively, starting with allele 1.
This format is convenient for markers with a small number of alleles.
- Extended format
- Lines in the allele frequency list are labelled A and consist of
a numeric allele label followed by an allele frequency. Alleles that
are not specifically listed are assumed to have frequency zero.
Classic Allele Frequency Format
For example, if some_marker has four alleles with frequencies 0.1,
0.2, 0.3 and 0.4 respectively and another_marker has two alleles with
frequencies 0.6 and 0.4 this is what the file might look like:
<contents of basic2.freq>
M some_marker
F 0.1 0.2 0.3 0.4
M another_marker
F 0.6 0.4
<end of basic2.freq>
An equivalent layout for the same information is:
<contents of basic2.freq>
M some_marker
F 0.1
F 0.2
F 0.3
F 0.4
M another_marker
F 0.6
F 0.4
<end of basic2.freq>
Extended allele frequency format
This format is recommended for microsatellites and other markers with large allele numbers.
For example, if you are analysing a microsatellite marker with alleles of size 152, 154 and 156
base-pairs and their respective frequencies are 0.5, 0.4 and 0.1 your frequency file might
read:
<contents of allele frequency file>
M some_microsatellite
A 152 0.5
A 154 0.4
A 156 0.1
<end of allele frequency file>
Well that is all you need to know about file formats to get started! You can proceed
to linkage analysis, ibd and
kinship estimation, haplotyping,
error detection or simulation. Have fun!
| 