| |
PSEUDO -- The Replicate Pool Method
The problem
Once a genome scan is performed, it is common practice to
determine significance of lod score peaks using simulation, specifically by
repeated linkage analysis on thousands of datasets generated by
gene-dropping to reproduce the underlying null distribution. This empirical
distribution can be used to estimate how frequently peaks of similar or greater
magnitude occur by chance in data of similar structure. One limitation of this
approach is the fact that individual simulations may take hours or days of
computational time to complete, making it impractical to perform enough simulations
to adequately evaluate significant findings.
How PSEUDO works
PSEUDO estimates p-values using the replicate pool method instead of the traditional
gene-dropping method for estimation of p-values . Instead of
recalculating
family-specific z-scores for each new
simulation, PSEUDO generates pseudosimulations by
resampling from a modest pool of pre-calculated values. One z-score replicate is chosen for each
family, and a Kong and Cox lod score
is generated by re-maximizing over delta. Because the most time-consuming portion of the overall lod score
calculation is the calculation of family-specific z-scores, PSEUDO is able to evaluate empirical p-values much more
efficiently than traditional methods. Depending on your data set, you could save hours or weeks of computational time.
Basic approach
The figure below illustrates the
basic approach for the replicate pool method for a simple scenario with four families (F1, F2,.
F3, F4) and a single analysis position.
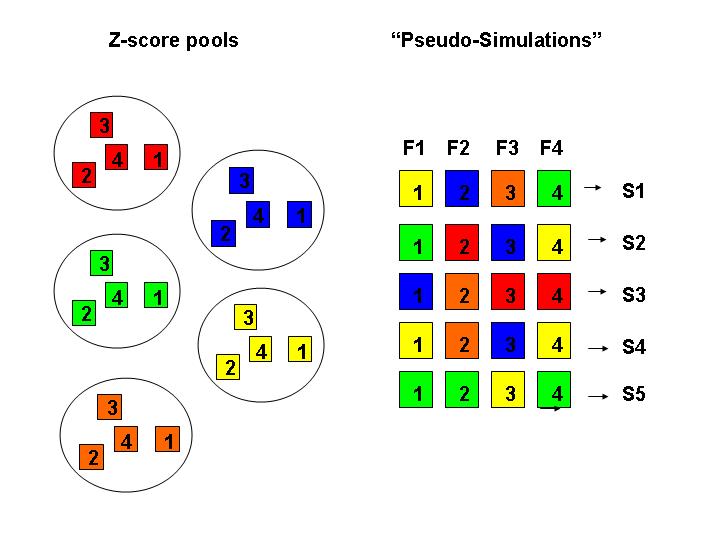
To begin, a reasonable number of pools (here we use five), each containing one pre-calculated z-score for each
family are generated.
"Pseudo-simulations"
S1 = (L01)
S2 = (L02)
S3 = (L03)
.
.
.
are generated by a selecting one zscore replicate for each family and calculating the Kong and Cox lod by summing
family z-scores at position 0 and remaximizing over delta.
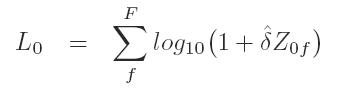
Multiple analysis positions
In practice, we consider p analysis positions located on C chromosomes. In this scenario, each pool contains one z-score for each family and analysis position.
Here, the basic sampling unit is the block Bcf of z-scores for family f from all
positions on chromosome c.
Additional "pseudosimulations" are generated by sampling one block of z-scores for each family and chromosome and recalculating the Kong and Cox lod at each position.
Each "pseudosimulation" Si replicates a genome scan with profile of p simulated lod scores
S1 = (L01, L11,... Lp1)
S2 = (L02, L12,... Lp2)
S3 = (L03, L13,... Lp3)
.
.
.
References
Song KK, Weeks DE, Sobel E, Feingold E (2004) Efficient Simulation of P Values for Linkage Analysis. Genetic
Epidemiology 24:1-9
Wigginton JE and Abecasis GR (2005) An Evaluation of the Replicate Pool Method : A Method for Quick Estimation of
Genomewide Linkage Peak P-Values. ...
Kong and Cox (1997) Allele-sharing models: LOD scores and accurate linkage tests. American Journal of Human
Genetics 61:1179-1188
Whittemore and Halpern (1994) A class of tests for linkage using affected pedigree members. Biometrics 50118-127
| |
