|
libStatGen Software
1
|
|
libStatGen Software
1
|
Allows the user to easily read/write a SAM/BAM file. More...
#include <SamFile.h>
Public Types | |
| enum | OpenType { READ, WRITE } |
| Enum for indicating whether to open the file for read or write. More... | |
| enum | SortedType { UNSORTED = 0, FLAG, COORDINATE, QUERY_NAME } |
| Enum for indicating the type of sort expected in the file. More... | |
Public Member Functions | |
| SamFile () | |
| Default Constructor, initializes the variables, but does not open any files. | |
| SamFile (ErrorHandler::HandlingType errorHandlingType) | |
| Constructor that sets the error handling type. | |
| SamFile (const char *filename, OpenType mode) | |
| Constructor that opens the specified file based on the specified mode (READ/WRITE), aborts if the file could not be opened. | |
| SamFile (const char *filename, OpenType mode, ErrorHandler::HandlingType errorHandlingType) | |
| Constructor that opens the specified file based on the specified mode (READ/WRITE) and handles errors per the specified handleType. | |
| SamFile (const char *filename, OpenType mode, SamFileHeader *header) | |
| Constructor that opens the specified file based on the specified mode (READ/WRITE) and reads the header, aborts if the file could not be opened or the header not read. | |
| SamFile (const char *filename, OpenType mode, ErrorHandler::HandlingType errorHandlingType, SamFileHeader *header) | |
| Constructor that opens the specified file based on the specified mode (READ/WRITE) and reads the header, handling errors per the specified handleType. | |
| virtual | ~SamFile () |
| Destructor. | |
| bool | OpenForRead (const char *filename, SamFileHeader *header=NULL) |
| Open a sam/bam file for reading with the specified filename, determing the type of file and SAM/BAM by reading the file (if not stdin). | |
| bool | OpenForWrite (const char *filename, SamFileHeader *header=NULL) |
| Open a sam/bam file for writing with the specified filename, determining SAM/BAM from the extension (.bam = BAM). | |
| bool | ReadBamIndex (const char *filename) |
| Read the specified bam index file. | |
| bool | ReadBamIndex () |
| Read the bam index file using the BAM filename as a base. | |
| void | SetReference (GenomeSequence *reference) |
| Sets the reference to the specified genome sequence object. | |
| void | SetReadSequenceTranslation (SamRecord::SequenceTranslation translation) |
| Set the type of sequence translation to use when reading the sequence. | |
| void | SetWriteSequenceTranslation (SamRecord::SequenceTranslation translation) |
| Set the type of sequence translation to use when writing the sequence. | |
| void | Close () |
| Close the file if there is one open. | |
| bool | IsOpen () |
| Returns whether or not the file has been opened successfully. | |
| bool | IsEOF () |
| Returns whether or not the end of the file has been reached. | |
| bool | ReadHeader (SamFileHeader &header) |
| Reads the header section from the file and stores it in the passed in header. | |
| bool | WriteHeader (SamFileHeader &header) |
| Writes the specified header into the file. | |
| bool | ReadRecord (SamFileHeader &header, SamRecord &record) |
| Reads the next record from the file & stores it in the passed in record. | |
| bool | WriteRecord (SamFileHeader &header, SamRecord &record) |
| Writes the specified record into the file. | |
| void | setSortedValidation (SortedType sortType) |
| Set the flag to validate that the file is sorted as it is read/written. | |
| uint32_t | GetCurrentRecordCount () |
| Return the number of records that have been read/written so far. | |
| SamStatus::Status | GetFailure () |
| Deprecated, get the Status of the last call that sets status. | |
| SamStatus::Status | GetStatus () |
| Get the Status of the last call that sets status. | |
| const char * | GetStatusMessage () |
| Get the Status Message of the last call that sets status. | |
| bool | SetReadSection (int32_t refID) |
| Sets which reference id (index into the BAM list of reference information) of the BAM file should be read. | |
| bool | SetReadSection (const char *refName) |
| Sets which reference name of the BAM file should be read. | |
| bool | SetReadSection (int32_t refID, int32_t start, int32_t end, bool overlap=true) |
| Sets which reference id (index into the BAM list of reference information) & start/end positions of the BAM file should be read. | |
| bool | SetReadSection (const char *refName, int32_t start, int32_t end, bool overlap=true) |
| Sets which reference name & start/end positions of the BAM file should be read. | |
| void | SetReadFlags (uint16_t requiredFlags, uint16_t excludedFlags) |
| Specify which reads should be returned by ReadRecord. | |
| int32_t | getNumMappedReadsFromIndex (int32_t refID) |
| Get the number of mapped reads in the specified reference id. | |
| int32_t | getNumUnMappedReadsFromIndex (int32_t refID) |
| Get the number of unmapped reads in the specified reference id. | |
| int32_t | getNumMappedReadsFromIndex (const char *refName, SamFileHeader &header) |
| Get the number of mapped reads in the specified reference name. | |
| int32_t | getNumUnMappedReadsFromIndex (const char *refName, SamFileHeader &header) |
| Get the number of unmapped reads in the specified reference name. | |
| uint32_t | GetNumOverlaps (SamRecord &samRecord) |
| Returns the number of bases in the passed in read that overlap the region that is currently set. | |
| void | GenerateStatistics (bool genStats) |
| Whether or not statistics should be generated for this file. | |
| const BamIndex * | GetBamIndex () |
| Return the bam index if one has been opened. | |
| int64_t | GetCurrentPosition () |
| Get the current file position. | |
| void | DisableBuffering () |
| Turn off file read buffering. | |
| void | PrintStatistics () |
| Print the statistics that have been recorded due to a call to GenerateStatistics. | |
| bool | attemptRecoverySync (bool(*checkSignature)(void *data), int length) |
| void | setAttemptRecovery (bool flag=false) |
Protected Member Functions | |
| void | init () |
| void | init (const char *filename, OpenType mode, SamFileHeader *header) |
| void | resetFile () |
| Resets the file prepping for a new file. | |
| bool | validateSortOrder (SamRecord &record, SamFileHeader &header) |
| Validate that the record is sorted compared to the previously read record if there is one, according to the specified sort order. | |
| SortedType | getSortOrderFromHeader (SamFileHeader &header) |
| bool | processNewSection (SamFileHeader &header) |
| bool | ensureIndexedReadPosition () |
| bool | checkRecordInSection (SamRecord &record) |
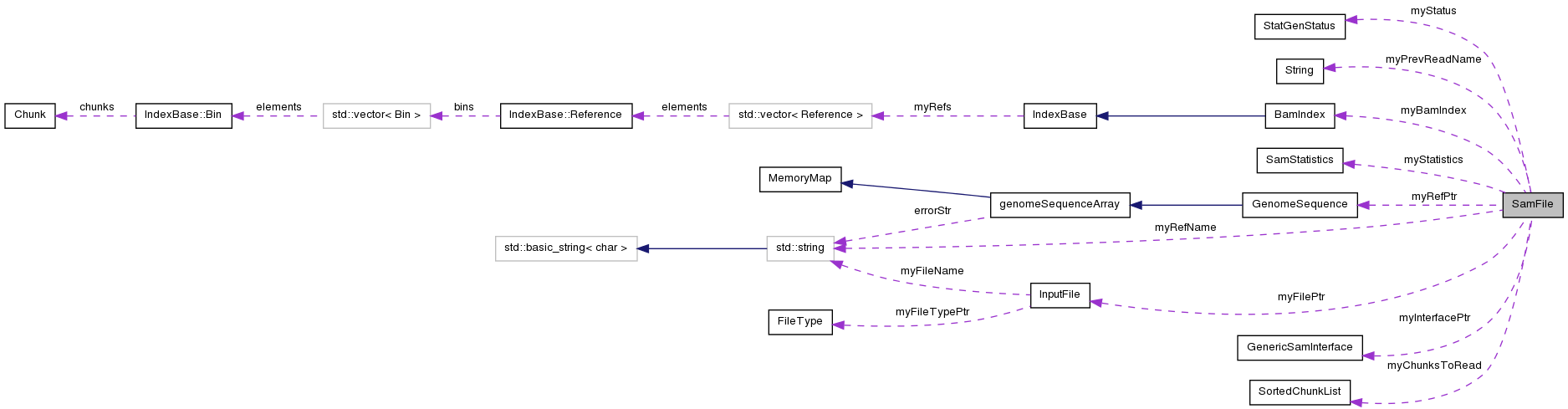
Protected Attributes | |
| IFILE | myFilePtr |
| GenericSamInterface * | myInterfacePtr |
| bool | myIsOpenForRead |
| Flag to indicate if a file is open for reading. | |
| bool | myIsOpenForWrite |
| Flag to indicate if a file is open for writing. | |
| bool | myHasHeader |
| Flag to indicate if a header has been read/written - required before being able to read/write a record. | |
| SortedType | mySortedType |
| int32_t | myPrevCoord |
| Previous values used for checking if the file is sorted. | |
| int32_t | myPrevRefID |
| String | myPrevReadName |
| uint32_t | myRecordCount |
| Keep a count of the number of records that have been read/written so far. | |
| SamStatistics * | myStatistics |
| Pointer to the statistics for this file. | |
| SamStatus | myStatus |
| The status of the last SamFile command. | |
| bool | myIsBamOpenForRead |
| Values for reading Sorted BAM files via the index. | |
| bool | myNewSection |
| bool | myOverlapSection |
| int32_t | myRefID |
| int32_t | myStartPos |
| int32_t | myEndPos |
| uint64_t | myCurrentChunkEnd |
| SortedChunkList | myChunksToRead |
| BamIndex * | myBamIndex |
| GenomeSequence * | myRefPtr |
| SamRecord::SequenceTranslation | myReadTranslation |
| SamRecord::SequenceTranslation | myWriteTranslation |
| std::string | myRefName |
Allows the user to easily read/write a SAM/BAM file.
The SamFile class contains additional functionality that allows a user to read specific sections of sorted & indexed BAM files. In order to take advantage of this capability, the index file must be read prior to setting the read section. This logic saves the time of having to read the entire file and takes advantage of the seeking capability of BGZF.
| enum SamFile::OpenType |
| enum SamFile::SortedType |
Enum for indicating the type of sort expected in the file.
| UNSORTED |
file is not sorted. |
| FLAG |
SO flag from the header indicates the sort type. |
| COORDINATE |
file is sorted by coordinate. |
| QUERY_NAME |
file is sorted by queryname. |
Definition at line 46 of file SamFile.h.
{
UNSORTED = 0, ///< file is not sorted.
FLAG, ///< SO flag from the header indicates the sort type.
COORDINATE, ///< file is sorted by coordinate.
QUERY_NAME ///< file is sorted by queryname.
};
| SamFile::SamFile | ( | ) |
Default Constructor, initializes the variables, but does not open any files.
Definition at line 26 of file SamFile.cpp.
References resetFile().
| SamFile::SamFile | ( | ErrorHandler::HandlingType | errorHandlingType | ) |
Constructor that sets the error handling type.
| errorHandlingType | how to handle errors. |
Definition at line 35 of file SamFile.cpp.
References resetFile().
| SamFile::SamFile | ( | const char * | filename, |
| OpenType | mode | ||
| ) |
Constructor that opens the specified file based on the specified mode (READ/WRITE), aborts if the file could not be opened.
| filename | name of the file to open. |
| mode | mode to use for opening the file. |
Definition at line 45 of file SamFile.cpp.
: myStatus() { init(filename, mode, NULL); }
| SamFile::SamFile | ( | const char * | filename, |
| OpenType | mode, | ||
| ErrorHandler::HandlingType | errorHandlingType | ||
| ) |
Constructor that opens the specified file based on the specified mode (READ/WRITE) and handles errors per the specified handleType.
| filename | name of the file to open. |
| mode | mode to use for opening the file. |
| errorHandlingType | how to handle errors. |
Definition at line 54 of file SamFile.cpp.
: myStatus(errorHandlingType) { init(filename, mode, NULL); }
| SamFile::SamFile | ( | const char * | filename, |
| OpenType | mode, | ||
| SamFileHeader * | header | ||
| ) |
Constructor that opens the specified file based on the specified mode (READ/WRITE) and reads the header, aborts if the file could not be opened or the header not read.
| filename | name of the file to open. |
| mode | mode to use for opening the file. |
| header | to read into or write from |
Definition at line 64 of file SamFile.cpp.
: myStatus() { init(filename, mode, header); }
| SamFile::SamFile | ( | const char * | filename, |
| OpenType | mode, | ||
| ErrorHandler::HandlingType | errorHandlingType, | ||
| SamFileHeader * | header | ||
| ) |
Constructor that opens the specified file based on the specified mode (READ/WRITE) and reads the header, handling errors per the specified handleType.
| filename | name of the file to open. |
| mode | mode to use for opening the file. |
| errorHandlingType | how to handle errors. |
| header | to read into or write from |
Definition at line 73 of file SamFile.cpp.
: myStatus(errorHandlingType) { init(filename, mode, header); }
| void SamFile::GenerateStatistics | ( | bool | genStats | ) |
Whether or not statistics should be generated for this file.
The value is carried over between files and is not reset, but the statistics themselves are reset between files.
| genStats | set to true if statistics should be generated, false if not. |
Definition at line 878 of file SamFile.cpp.
References myStatistics.
{
if(genStats)
{
if(myStatistics == NULL)
{
// Want to generate statistics, but do not yet have the
// structure for them, so create one.
myStatistics = new SamStatistics();
}
}
else
{
// Do not generate statistics, so if myStatistics is not NULL,
// delete it.
if(myStatistics != NULL)
{
delete myStatistics;
myStatistics = NULL;
}
}
}
| const BamIndex * SamFile::GetBamIndex | ( | ) |
Return the bam index if one has been opened.
Definition at line 903 of file SamFile.cpp.
{
return(myBamIndex);
}
| int64_t SamFile::GetCurrentPosition | ( | ) | [inline] |
| SamStatus::Status SamFile::GetFailure | ( | ) | [inline] |
Deprecated, get the Status of the last call that sets status.
To remain backwards compatable - will be removed later.
Definition at line 201 of file SamFile.h.
References GetStatus().
{
return(GetStatus());
}
| int32_t SamFile::getNumMappedReadsFromIndex | ( | int32_t | refID | ) |
Get the number of mapped reads in the specified reference id.
Returns -1 for out of range refIDs.
| refID | reference ID for which to extract the number of mapped reads. |
Definition at line 790 of file SamFile.cpp.
References StatGenStatus::FAIL_ORDER, BamIndex::getNumMappedReads(), myStatus, and StatGenStatus::setStatus().
{
// The bam index must have already been read.
if(myBamIndex == NULL)
{
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot get num mapped reads from the index until it has been read.");
return(false);
}
return(myBamIndex->getNumMappedReads(refID));
}
| int32_t SamFile::getNumMappedReadsFromIndex | ( | const char * | refName, |
| SamFileHeader & | header | ||
| ) |
Get the number of mapped reads in the specified reference name.
Returns -1 for unknown reference names.
| refName | reference name for which to extract the number of mapped reads. |
| header | header object containing the map from refName to refID |
Definition at line 820 of file SamFile.cpp.
References StatGenStatus::FAIL_ORDER, BamIndex::getNumMappedReads(), SamFileHeader::getReferenceID(), myStatus, BamIndex::REF_ID_UNMAPPED, and StatGenStatus::setStatus().
{
// The bam index must have already been read.
if(myBamIndex == NULL)
{
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot get num mapped reads from the index until it has been read.");
return(false);
}
int32_t refID = BamIndex::REF_ID_UNMAPPED;
if((strcmp(refName, "") != 0) && (strcmp(refName, "*") != 0))
{
// Reference name specified, so read just the "-1" entries.
refID = header.getReferenceID(refName);
}
return(myBamIndex->getNumMappedReads(refID));
}
| uint32_t SamFile::GetNumOverlaps | ( | SamRecord & | samRecord | ) |
Returns the number of bases in the passed in read that overlap the region that is currently set.
Overlapping means that the bases occur in both the read and the reference as either matches or mismatches. This does not count insertions, deletions, clips, pads, or skips.
| samRecord | to check for overlapping bases. |
Definition at line 864 of file SamFile.cpp.
References SamRecord::getNumOverlaps(), SamRecord::setReference(), and SamRecord::setSequenceTranslation().
{
if(myRefPtr != NULL)
{
samRecord.setReference(myRefPtr);
}
samRecord.setSequenceTranslation(myReadTranslation);
// Get the overlaps in the sam record for the region currently set
// for this file.
return(samRecord.getNumOverlaps(myStartPos, myEndPos));
}
| int32_t SamFile::getNumUnMappedReadsFromIndex | ( | int32_t | refID | ) |
Get the number of unmapped reads in the specified reference id.
Returns -1 for out of range refIDs.
| refID | reference ID for which to extract the number of unmapped reads. |
Definition at line 805 of file SamFile.cpp.
References StatGenStatus::FAIL_ORDER, BamIndex::getNumUnMappedReads(), myStatus, and StatGenStatus::setStatus().
{
// The bam index must have already been read.
if(myBamIndex == NULL)
{
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot get num unmapped reads from the index until it has been read.");
return(false);
}
return(myBamIndex->getNumUnMappedReads(refID));
}
| int32_t SamFile::getNumUnMappedReadsFromIndex | ( | const char * | refName, |
| SamFileHeader & | header | ||
| ) |
Get the number of unmapped reads in the specified reference name.
Returns -1 for unknown reference names.
| refName | reference name for which to extract the number of unmapped reads. |
| header | header object containing the map from refName to refID |
Definition at line 842 of file SamFile.cpp.
References StatGenStatus::FAIL_ORDER, BamIndex::getNumUnMappedReads(), SamFileHeader::getReferenceID(), myStatus, BamIndex::REF_ID_UNMAPPED, and StatGenStatus::setStatus().
{
// The bam index must have already been read.
if(myBamIndex == NULL)
{
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot get num unmapped reads from the index until it has been read.");
return(false);
}
int32_t refID = BamIndex::REF_ID_UNMAPPED;
if((strcmp(refName, "") != 0) && (strcmp(refName, "*") != 0))
{
// Reference name specified, so read just the "-1" entries.
refID = header.getReferenceID(refName);
}
return(myBamIndex->getNumUnMappedReads(refID));
}
| bool SamFile::IsEOF | ( | ) |
Returns whether or not the end of the file has been reached.
Definition at line 424 of file SamFile.cpp.
References ifeof().
{
if (myFilePtr != NULL)
{
// File Pointer is set, so return if eof.
return(ifeof(myFilePtr));
}
// File pointer is not set, so return true, eof.
return true;
}
| bool SamFile::IsOpen | ( | ) |
Returns whether or not the file has been opened successfully.
Definition at line 410 of file SamFile.cpp.
References InputFile::isOpen().
{
if (myFilePtr != NULL)
{
// File Pointer is set, so return if it is open.
return(myFilePtr->isOpen());
}
// File pointer is not set, so return false, not open.
return false;
}
| bool SamFile::OpenForRead | ( | const char * | filename, |
| SamFileHeader * | header = NULL |
||
| ) |
Open a sam/bam file for reading with the specified filename, determing the type of file and SAM/BAM by reading the file (if not stdin).
| filename | the sam/bam file to open for reading. |
| header | to read into or write from (optional) |
Definition at line 93 of file SamFile.cpp.
References InputFile::BGZF, InputFile::DEFAULT, StatGenStatus::FAIL_IO, ifopen(), ifread(), ifrewind(), myIsBamOpenForRead, myIsOpenForRead, myStatus, ReadHeader(), resetFile(), InputFile::setAttemptRecovery(), StatGenStatus::setStatus(), StatGenStatus::SUCCESS, and InputFile::UNCOMPRESSED.
Referenced by Pileup< PILEUP_TYPE, FUNC_CLASS >::processFile().
{
// Reset for any previously operated on files.
resetFile();
int lastchar = 0;
while (filename[lastchar] != 0) lastchar++;
// If at least one character, check for '-'.
if((lastchar >= 1) && (filename[0] == '-'))
{
// Read from stdin - determine type of file to read.
// Determine if compressed bam.
if(strcmp(filename, "-.bam") == 0)
{
// Compressed bam - open as bgzf.
// -.bam is the filename, read compressed bam from stdin
filename = "-";
myFilePtr = new InputFile;
// support recover mode - this switches in a reader
// capable of recovering from bad BGZF compression blocks.
myFilePtr->setAttemptRecovery(myAttemptRecovery);
myFilePtr->openFile(filename, "rb", InputFile::BGZF);
myInterfacePtr = new BamInterface;
// Read the magic string.
char magic[4];
ifread(myFilePtr, magic, 4);
}
else if(strcmp(filename, "-.ubam") == 0)
{
// uncompressed BAM File.
// -.ubam is the filename, read uncompressed bam from stdin.
// uncompressed BAM is still compressed with BGZF, but using
// compression level 0, so still open as BGZF since it has a
// BGZF header.
filename = "-";
// Uncompressed, so do not require the eof block.
#ifdef __ZLIB_AVAILABLE__
BgzfFileType::setRequireEofBlock(false);
#endif
myFilePtr = ifopen(filename, "rb", InputFile::BGZF);
myInterfacePtr = new BamInterface;
// Read the magic string.
char magic[4];
ifread(myFilePtr, magic, 4);
}
else if((strcmp(filename, "-") == 0) || (strcmp(filename, "-.sam") == 0))
{
// SAM File.
// read sam from stdin
filename = "-";
myFilePtr = ifopen(filename, "rb", InputFile::UNCOMPRESSED);
myInterfacePtr = new SamInterface;
}
else
{
std::string errorMessage = "Invalid SAM/BAM filename, ";
errorMessage += filename;
errorMessage += ". From stdin, can only be '-', '-.sam', '-.bam', or '-.ubam'";
myStatus.setStatus(SamStatus::FAIL_IO, errorMessage.c_str());
delete myFilePtr;
myFilePtr = NULL;
return(false);
}
}
else
{
// Not from stdin. Read the file to determine the type.
myFilePtr = new InputFile;
// support recovery mode - this conditionally enables a reader
// capable of recovering from bad BGZF compression blocks.
myFilePtr->setAttemptRecovery(myAttemptRecovery);
bool rc = myFilePtr->openFile(filename, "rb", InputFile::DEFAULT);
if (rc == false)
{
std::string errorMessage = "Failed to Open ";
errorMessage += filename;
errorMessage += " for reading";
myStatus.setStatus(SamStatus::FAIL_IO, errorMessage.c_str());
delete myFilePtr;
myFilePtr = NULL;
return(false);
}
char magic[4];
ifread(myFilePtr, magic, 4);
if (magic[0] == 'B' && magic[1] == 'A' && magic[2] == 'M' &&
magic[3] == 1)
{
myInterfacePtr = new BamInterface;
// Set that it is a bam file open for reading. This is needed to
// determine if an index file can be used.
myIsBamOpenForRead = true;
}
else
{
// Not a bam, so rewind to the beginning of the file so it
// can be read.
ifrewind(myFilePtr);
myInterfacePtr = new SamInterface;
}
}
// File is open for reading.
myIsOpenForRead = true;
// Read the header if one was passed in.
if(header != NULL)
{
return(ReadHeader(*header));
}
// Successfully opened the file.
myStatus = SamStatus::SUCCESS;
return(true);
}
| bool SamFile::OpenForWrite | ( | const char * | filename, |
| SamFileHeader * | header = NULL |
||
| ) |
Open a sam/bam file for writing with the specified filename, determining SAM/BAM from the extension (.bam = BAM).
| filename | the sam/bam file to open for writing. |
| header | to read into or write from (optional) |
Definition at line 223 of file SamFile.cpp.
References InputFile::BGZF, StatGenStatus::FAIL_IO, ifopen(), myIsOpenForWrite, myStatus, resetFile(), StatGenStatus::setStatus(), StatGenStatus::SUCCESS, InputFile::UNCOMPRESSED, and WriteHeader().
{
// Reset for any previously operated on files.
resetFile();
int lastchar = 0;
while (filename[lastchar] != 0) lastchar++;
if (lastchar >= 4 &&
filename[lastchar - 4] == 'u' &&
filename[lastchar - 3] == 'b' &&
filename[lastchar - 2] == 'a' &&
filename[lastchar - 1] == 'm')
{
// BAM File.
// if -.ubam is the filename, write uncompressed bam to stdout
if((lastchar == 6) && (filename[0] == '-') && (filename[1] == '.'))
{
filename = "-";
}
myFilePtr = ifopen(filename, "wb0", InputFile::BGZF);
myInterfacePtr = new BamInterface;
}
else if (lastchar >= 3 &&
filename[lastchar - 3] == 'b' &&
filename[lastchar - 2] == 'a' &&
filename[lastchar - 1] == 'm')
{
// BAM File.
// if -.bam is the filename, write compressed bam to stdout
if((lastchar == 5) && (filename[0] == '-') && (filename[1] == '.'))
{
filename = "-";
}
myFilePtr = ifopen(filename, "wb", InputFile::BGZF);
myInterfacePtr = new BamInterface;
}
else
{
// SAM File
// if - (followed by anything is the filename,
// write uncompressed sam to stdout
if((lastchar >= 1) && (filename[0] == '-'))
{
filename = "-";
}
myFilePtr = ifopen(filename, "wb", InputFile::UNCOMPRESSED);
myInterfacePtr = new SamInterface;
}
if (myFilePtr == NULL)
{
std::string errorMessage = "Failed to Open ";
errorMessage += filename;
errorMessage += " for writing";
myStatus.setStatus(SamStatus::FAIL_IO, errorMessage.c_str());
return(false);
}
myIsOpenForWrite = true;
// Write the header if one was passed in.
if(header != NULL)
{
return(WriteHeader(*header));
}
// Successfully opened the file.
myStatus = SamStatus::SUCCESS;
return(true);
}
| void SamFile::PrintStatistics | ( | ) | [inline] |
Print the statistics that have been recorded due to a call to GenerateStatistics.
Definition at line 352 of file SamFile.h.
References myStatistics.
{if(myStatistics != NULL) myStatistics->print();}
| bool SamFile::ReadBamIndex | ( | const char * | filename | ) |
Read the specified bam index file.
It must be read prior to setting a read section, for seeking and reading portions of a bam file.
| filename | the name of the bam index file to be read. |
Definition at line 300 of file SamFile.cpp.
References myStatus, BamIndex::readIndex(), StatGenStatus::setStatus(), and StatGenStatus::SUCCESS.
{
// Cleanup a previously setup index.
if(myBamIndex != NULL)
{
delete myBamIndex;
myBamIndex = NULL;
}
// Create a new bam index.
myBamIndex = new BamIndex();
SamStatus::Status indexStat = myBamIndex->readIndex(bamIndexFilename);
if(indexStat != SamStatus::SUCCESS)
{
std::string errorMessage = "Failed to read the bam Index file: ";
errorMessage += bamIndexFilename;
myStatus.setStatus(indexStat, errorMessage.c_str());
delete myBamIndex;
myBamIndex = NULL;
return(false);
}
myStatus = SamStatus::SUCCESS;
return(true);
}
| bool SamFile::ReadBamIndex | ( | ) |
Read the bam index file using the BAM filename as a base.
It must be read prior to setting a read section, for seeking and reading portions of a bam file. Must be read after opening the BAM file since it uses the BAM filename as a base name for the index file. First it tries filename.bam.bai. If that fails, it tries it without the .bam extension, filename.bai.
Definition at line 328 of file SamFile.cpp.
References StatGenStatus::FAIL_ORDER, InputFile::getFileName(), myStatus, and StatGenStatus::setStatus().
{
if(myFilePtr == NULL)
{
// Can't read the bam index file because the BAM file has not yet been
// opened, so we don't know the base filename for the index file.
std::string errorMessage = "Failed to read the bam Index file -"
" the BAM file needs to be read first in order to determine"
" the index filename.";
myStatus.setStatus(SamStatus::FAIL_ORDER, errorMessage.c_str());
return(false);
}
const char* bamBaseName = myFilePtr->getFileName();
std::string indexName = bamBaseName;
indexName += ".bai";
bool foundFile = true;
try
{
if(ReadBamIndex(indexName.c_str()) == false)
{
foundFile = false;
}
}
catch (std::exception&)
{
foundFile = false;
}
// Check to see if the index file was found.
if(!foundFile)
{
// Not found - try without the bam extension.
// Locate the start of the bam extension
size_t startExt = indexName.find(".bam");
if(startExt == std::string::npos)
{
// Could not find the .bam extension, so just return false since the
// call to ReadBamIndex set the status.
return(false);
}
// Remove ".bam" and try reading the index again.
indexName.erase(startExt, 4);
return(ReadBamIndex(indexName.c_str()));
}
return(true);
}
| bool SamFile::ReadHeader | ( | SamFileHeader & | header | ) |
Reads the header section from the file and stores it in the passed in header.
Definition at line 437 of file SamFile.cpp.
References StatGenStatus::FAIL_ORDER, myHasHeader, myIsOpenForRead, myStatus, StatGenStatus::setStatus(), and StatGenStatus::SUCCESS.
Referenced by OpenForRead(), and Pileup< PILEUP_TYPE, FUNC_CLASS >::processFile().
{
myStatus = SamStatus::SUCCESS;
if(myIsOpenForRead == false)
{
// File is not open for read
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot read header since the file is not open for reading");
return(false);
}
if(myHasHeader == true)
{
// The header has already been read.
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot read header since it has already been read.");
return(false);
}
if(myInterfacePtr->readHeader(myFilePtr, header, myStatus))
{
// The header has now been successfully read.
myHasHeader = true;
return(true);
}
return(false);
}
| bool SamFile::ReadRecord | ( | SamFileHeader & | header, |
| SamRecord & | record | ||
| ) |
Reads the next record from the file & stores it in the passed in record.
If it is an indexed BAM file and SetReadSection was called, only alignments in the section specified by SetReadSection are read. If they all have already been read, this method returns false.
Validates that the record is sorted according to the value set by setSortedValidation. No sorting validation is done if specified to be unsorted, or setSortedValidation was never called.
Definition at line 501 of file SamFile.cpp.
References StatGenStatus::FAIL_ORDER, SamRecord::getFlag(), myHasHeader, myIsOpenForRead, myRecordCount, myStatistics, myStatus, SamRecord::setReference(), SamRecord::setSequenceTranslation(), StatGenStatus::setStatus(), StatGenStatus::SUCCESS, and validateSortOrder().
Referenced by Pileup< PILEUP_TYPE, FUNC_CLASS >::processFile().
{
myStatus = SamStatus::SUCCESS;
if(myIsOpenForRead == false)
{
// File is not open for read
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot read record since the file is not open for reading");
throw(std::runtime_error("SOFTWARE BUG: trying to read a SAM/BAM record prior to opening the file."));
return(false);
}
if(myHasHeader == false)
{
// The header has not yet been read.
// TODO - maybe just read the header.
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot read record since the header has not been read.");
throw(std::runtime_error("SOFTWARE BUG: trying to read a SAM/BAM record prior to reading the header."));
return(false);
}
// Check to see if a new region has been set. If so, determine the
// chunks for that region.
if(myNewSection)
{
if(!processNewSection(header))
{
// Failed processing a new section. Could be an
// order issue like the file not being open or the
// indexed file not having been read.
// processNewSection sets myStatus with the failure reason.
return(false);
}
}
// Read until a record is not successfully read or there are no more
// requested records.
while(myStatus == SamStatus::SUCCESS)
{
record.setReference(myRefPtr);
record.setSequenceTranslation(myReadTranslation);
// If reading by index, this method will setup to ensure it is in
// the correct position for the next record (if not already there).
// Sets myStatus if it could not move to a good section.
// Just returns true if it is not setup to read by index.
if(!ensureIndexedReadPosition())
{
// Either there are no more records in the section
// or it failed to move to the right section, so there
// is nothing more to read, stop looping.
break;
}
// File is open for reading and the header has been read, so read the
// next record.
myInterfacePtr->readRecord(myFilePtr, header, record, myStatus);
if(myStatus != SamStatus::SUCCESS)
{
// Failed to read the record, so break out of the loop.
break;
}
// Successfully read a record, so check if we should filter it.
// First check if it is out of the section. Returns true
// if not reading by sections, returns false if the record
// is outside of the section. Sets status to NO_MORE_RECS if
// there is nothing left ot read in the section.
if(!checkRecordInSection(record))
{
// The record is not in the section.
// The while loop will detect if NO_MORE_RECS was set.
continue;
}
// Check the flag for required/excluded flags.
uint16_t flag = record.getFlag();
if((flag & myRequiredFlags) != myRequiredFlags)
{
// The record does not conatain all required flags, so
// continue looking.
continue;
}
if((flag & myExcludedFlags) != 0)
{
// The record contains an excluded flag, so continue looking.
continue;
}
//increment the record count.
myRecordCount++;
if(myStatistics != NULL)
{
// Statistics should be updated.
myStatistics->updateStatistics(record);
}
// Successfully read the record, so check the sort order.
if(!validateSortOrder(record, header))
{
// ValidateSortOrder sets the status on a failure.
return(false);
}
return(true);
} // End while loop that checks if a desired record is found or failure.
// Return true if a record was found.
return(myStatus == SamStatus::SUCCESS);
}
| void SamFile::SetReadFlags | ( | uint16_t | requiredFlags, |
| uint16_t | excludedFlags | ||
| ) |
Specify which reads should be returned by ReadRecord.
Reads will only be returned by ReadRecord that contain the specified required flags and that do not contain any of the specified excluded flags. ReadRecord will continue to read from the file until a record that complies with these flag settings is found or until the end of the file/region.
| requiredFlags | flags that are required to be in records returned by ReadRecord (set to 0x0 if there are no required flags). |
| excludedFlags | flags that are required to not be in records returned by ReadRecord (set to 0x0 if there are no excluded flags). |
Definition at line 781 of file SamFile.cpp.
{
myRequiredFlags = requiredFlags;
myExcludedFlags = excludedFlags;
}
| bool SamFile::SetReadSection | ( | int32_t | refID | ) |
Sets which reference id (index into the BAM list of reference information) of the BAM file should be read.
The records for that reference id will be retrieved on each ReadRecord call. Reference ids start at 0, and -1 indicates reads with no reference. When all records have been retrieved for the specified reference id, ReadRecord will return failure until a new read section is set. Must be called only after the file has been opened for reading. Sorting validation is reset everytime SetReadPosition is called since it can jump around in the file.
| refID | the reference ID of the records to read from the file. |
Definition at line 683 of file SamFile.cpp.
Referenced by SetReadSection().
{
// No start/end specified, so set back to default -1.
return(SetReadSection(refID, -1, -1));
}
| bool SamFile::SetReadSection | ( | const char * | refName | ) |
Sets which reference name of the BAM file should be read.
The records for that reference name will be retrieved on each ReadRecord call. Specify "" or "*" to read records not associated with a reference. When all records have been retrieved for the specified reference name, ReadRecord will return failure until a new read section is set. Must be called only after the file has been opened for reading. Sorting validation is reset everytime SetReadPosition is called since it can jump around in the file.
| refName | the reference name of the records to read from the file. |
Definition at line 692 of file SamFile.cpp.
References SetReadSection().
{
// No start/end specified, so set back to default -1.
return(SetReadSection(refName, -1, -1));
}
| bool SamFile::SetReadSection | ( | int32_t | refID, |
| int32_t | start, | ||
| int32_t | end, | ||
| bool | overlap = true |
||
| ) |
Sets which reference id (index into the BAM list of reference information) & start/end positions of the BAM file should be read.
The records for that reference id and positions will be retrieved on each ReadRecord call. Reference ids start at 0, and -1 indicates reads with no reference. When all records have been retrieved for the specified reference id, ReadRecord will return failure until a new read section is set. Must be called only after the file has been opened for reading. Sorting validation is reset everytime SetReadPosition is called since it can jump around in the file.
| refID | the reference ID of the records to read from the file. |
| start | inclusive 0-based start position of records that should be read for this refID. |
| end | exclusive 0-based end position of records that should be read for this refID. |
| overlap | When true (default), return reads that just overlap the region; when false, only return reads that fall completely within the region |
Definition at line 700 of file SamFile.cpp.
References StatGenStatus::FAIL_ORDER, myIsBamOpenForRead, myPrevCoord, myStatus, StatGenStatus::setStatus(), and StatGenStatus::SUCCESS.
{
// If there is not a BAM file open for reading, return failure.
// Opening a new file clears the read section, so it must be
// set after the file is opened.
if(!myIsBamOpenForRead)
{
// There is not a BAM file open for reading.
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot set section since there is no bam file open");
return(false);
}
myNewSection = true;
myOverlapSection = overlap;
myStartPos = start;
myEndPos = end;
myRefID = refID;
myRefName.clear();
myChunksToRead.clear();
// Reset the end of the current chunk. We are resetting our read, so
// we no longer have a "current chunk" that we are reading.
myCurrentChunkEnd = 0;
myStatus = SamStatus::SUCCESS;
// Reset the sort order criteria since we moved around in the file.
myPrevCoord = -1;
myPrevRefID = 0;
myPrevReadName.Clear();
return(true);
}
| bool SamFile::SetReadSection | ( | const char * | refName, |
| int32_t | start, | ||
| int32_t | end, | ||
| bool | overlap = true |
||
| ) |
Sets which reference name & start/end positions of the BAM file should be read.
The records for this reference name & positions will be retrieved on each ReadRecord call. Specify "" or "*" to indicate reads with no reference. When all records have been retrieved for the specified section, ReadRecord will return failure until a new read section is set. Must be called only after the file has been opened for reading. Sorting validation is reset everytime SetReadSection is called since it can jump around in the file.
| refName | the reference name of the records to read from the file. |
| start | inclusive 0-based start position of records that should be read for this refID. |
| end | exclusive 0-based end position of records that should be read for this refID. |
| overlap | When true (default), return reads that just overlap the region; when false, only return reads that fall completely within the region |
Definition at line 736 of file SamFile.cpp.
References StatGenStatus::FAIL_ORDER, myIsBamOpenForRead, myPrevCoord, myStatus, BamIndex::REF_ID_ALL, BamIndex::REF_ID_UNMAPPED, StatGenStatus::setStatus(), and StatGenStatus::SUCCESS.
{
// If there is not a BAM file open for reading, return failure.
// Opening a new file clears the read section, so it must be
// set after the file is opened.
if(!myIsBamOpenForRead)
{
// There is not a BAM file open for reading.
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot set section since there is no bam file open");
return(false);
}
myNewSection = true;
myOverlapSection = overlap;
myStartPos = start;
myEndPos = end;
if((strcmp(refName, "") == 0) || (strcmp(refName, "*") == 0))
{
// No Reference name specified, so read just the "-1" entries.
myRefID = BamIndex::REF_ID_UNMAPPED;
}
else
{
// save the reference name and revert the reference ID to unknown
// so it will be calculated later.
myRefName = refName;
myRefID = BamIndex::REF_ID_ALL;
}
myChunksToRead.clear();
// Reset the end of the current chunk. We are resetting our read, so
// we no longer have a "current chunk" that we are reading.
myCurrentChunkEnd = 0;
myStatus = SamStatus::SUCCESS;
// Reset the sort order criteria since we moved around in the file.
myPrevCoord = -1;
myPrevRefID = 0;
myPrevReadName.Clear();
return(true);
}
| void SamFile::SetReadSequenceTranslation | ( | SamRecord::SequenceTranslation | translation | ) |
Set the type of sequence translation to use when reading the sequence.
Passed down to the SamRecord when it is read. The default type (if this method is never called) is NONE (the sequence is left as-is).
| translation | type of sequence translation to use. |
Definition at line 387 of file SamFile.cpp.
{
myReadTranslation = translation;
}
| void SamFile::SetReference | ( | GenomeSequence * | reference | ) |
Sets the reference to the specified genome sequence object.
| reference | pointer to the GenomeSequence object. |
Definition at line 380 of file SamFile.cpp.
Referenced by Pileup< PILEUP_TYPE, FUNC_CLASS >::processFile().
{
myRefPtr = reference;
}
| void SamFile::setSortedValidation | ( | SortedType | sortType | ) |
Set the flag to validate that the file is sorted as it is read/written.
Must be called after the file has been opened. Sorting validation is reset everytime SetReadPosition is called since it can jump around in the file.
| sortType | specifies the type of sort to be checked for. |
Definition at line 669 of file SamFile.cpp.
Referenced by Pileup< PILEUP_TYPE, FUNC_CLASS >::processFile().
{
mySortedType = sortType;
}
| void SamFile::SetWriteSequenceTranslation | ( | SamRecord::SequenceTranslation | translation | ) |
Set the type of sequence translation to use when writing the sequence.
Passed down to the SamRecord when it is written. The default type (if this method is never called) is NONE (the sequence is left as-is).
| translation | type of sequence translation to use. |
Definition at line 394 of file SamFile.cpp.
{
myWriteTranslation = translation;
}
| bool SamFile::validateSortOrder | ( | SamRecord & | record, |
| SamFileHeader & | header | ||
| ) | [protected] |
Validate that the record is sorted compared to the previously read record if there is one, according to the specified sort order.
If the sort order is UNSORTED, true is returned. Sorting validation is reset everytime SetReadPosition is called since it can jump around in the file.
Definition at line 1006 of file SamFile.cpp.
References FLAG, SamRecord::get0BasedPosition(), SamRecord::getReadName(), SamRecord::getReferenceID(), SamFileHeader::getReferenceLabel(), StatGenStatus::INVALID_SORT, myPrevCoord, myRecordCount, myStatus, QUERY_NAME, BamIndex::REF_ID_UNMAPPED, SamRecord::setReference(), SamRecord::setSequenceTranslation(), StatGenStatus::setStatus(), and UNSORTED.
Referenced by ReadRecord(), and WriteRecord().
{
if(myRefPtr != NULL)
{
record.setReference(myRefPtr);
}
record.setSequenceTranslation(myReadTranslation);
bool status = false;
if(mySortedType == UNSORTED)
{
// Unsorted, so nothing to validate, just return true.
status = true;
}
else
{
// Check to see if mySortedType is based on the header.
if(mySortedType == FLAG)
{
// Determine the sorted type from what was read out of the header.
mySortedType = getSortOrderFromHeader(header);
}
if(mySortedType == QUERY_NAME)
{
// Validate that it is sorted by query name.
// Get the query name from the record.
const char* readName = record.getReadName();
// Check if it is sorted either in samtools way or picard's way.
if((myPrevReadName.Compare(readName) > 0) &&
(strcmp(myPrevReadName.c_str(), readName) > 0))
{
// return false.
String errorMessage = "ERROR: File is not sorted by read name at record ";
errorMessage += myRecordCount;
errorMessage += "\n\tPrevious record was ";
errorMessage += myPrevReadName;
errorMessage += ", but this record is ";
errorMessage += readName;
myStatus.setStatus(SamStatus::INVALID_SORT,
errorMessage.c_str());
status = false;
}
else
{
myPrevReadName = readName;
status = true;
}
}
else
{
// Validate that it is sorted by COORDINATES.
// Get the leftmost coordinate and the reference index.
int32_t refID = record.getReferenceID();
int32_t coord = record.get0BasedPosition();
// The unmapped reference id is at the end of a sorted file.
if(refID == BamIndex::REF_ID_UNMAPPED)
{
// A new reference ID that is for the unmapped reads
// is always valid.
status = true;
myPrevRefID = refID;
myPrevCoord = coord;
}
else if(myPrevRefID == BamIndex::REF_ID_UNMAPPED)
{
// Previous reference ID was for unmapped reads, but the
// current one is not, so this is not sorted.
String errorMessage = "ERROR: File is not coordinate sorted at record ";
errorMessage += myRecordCount;
errorMessage += "\n\tPrevious record was unmapped, but this record is ";
errorMessage += header.getReferenceLabel(refID) + ":" + coord;
myStatus.setStatus(SamStatus::INVALID_SORT,
errorMessage.c_str());
status = false;
}
else if(refID < myPrevRefID)
{
// Current reference id is less than the previous one,
//meaning that it is not sorted.
String errorMessage = "ERROR: File is not coordinate sorted at record ";
errorMessage += myRecordCount;
errorMessage += "\n\tPrevious record was ";
errorMessage += header.getReferenceLabel(myPrevRefID) + ":" + myPrevCoord;
errorMessage += ", but this record is ";
errorMessage += header.getReferenceLabel(refID) + ":" + coord;
myStatus.setStatus(SamStatus::INVALID_SORT,
errorMessage.c_str());
status = false;
}
else
{
// The reference IDs are in the correct order.
if(refID > myPrevRefID)
{
// New reference id, so set the previous coordinate to -1
myPrevCoord = -1;
}
// Check the coordinates.
if(coord < myPrevCoord)
{
// New Coord is less than the previous position.
String errorMessage = "ERROR: File is not coordinate sorted at record ";
errorMessage += myRecordCount;
errorMessage += "\n\tPreviousRecord was ";
errorMessage += header.getReferenceLabel(myPrevRefID) + ":" + myPrevCoord;
errorMessage += ", but this record is ";
errorMessage += header.getReferenceLabel(refID) + ":" + coord;
myStatus.setStatus(SamStatus::INVALID_SORT,
errorMessage.c_str());
status = false;
}
else
{
myPrevRefID = refID;
myPrevCoord = coord;
status = true;
}
}
}
}
return(status);
}
| bool SamFile::WriteHeader | ( | SamFileHeader & | header | ) |
Writes the specified header into the file.
Definition at line 467 of file SamFile.cpp.
References StatGenStatus::FAIL_ORDER, myHasHeader, myIsOpenForWrite, myStatus, StatGenStatus::setStatus(), and StatGenStatus::SUCCESS.
Referenced by OpenForWrite().
{
myStatus = SamStatus::SUCCESS;
if(myIsOpenForWrite == false)
{
// File is not open for write
// -OR-
// The header has already been written.
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot write header since the file is not open for writing");
return(false);
}
if(myHasHeader == true)
{
// The header has already been written.
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot write header since it has already been written");
return(false);
}
if(myInterfacePtr->writeHeader(myFilePtr, header, myStatus))
{
// The header has now been successfully written.
myHasHeader = true;
return(true);
}
// return the status.
return(false);
}
| bool SamFile::WriteRecord | ( | SamFileHeader & | header, |
| SamRecord & | record | ||
| ) |
Writes the specified record into the file.
Validates that the record is sorted according to the value set by setSortedValidation. No sorting validation is done if specified to be unsorted, or setSortedValidation was never called. Returns false and does not write the record if the record was not properly sorted.
Definition at line 619 of file SamFile.cpp.
References StatGenStatus::FAIL_ORDER, StatGenStatus::INVALID_SORT, myHasHeader, myIsOpenForWrite, myRecordCount, myStatus, SamRecord::setReference(), StatGenStatus::setStatus(), StatGenStatus::SUCCESS, and validateSortOrder().
Referenced by SamCoordOutput::flush().
{
if(myIsOpenForWrite == false)
{
// File is not open for writing
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot write record since the file is not open for writing");
return(false);
}
if(myHasHeader == false)
{
// The header has not yet been written.
myStatus.setStatus(SamStatus::FAIL_ORDER,
"Cannot write record since the header has not been written");
return(false);
}
// Before trying to write the record, validate the sort order.
if(!validateSortOrder(record, header))
{
// Not sorted like it is supposed to be, do not write the record
myStatus.setStatus(SamStatus::INVALID_SORT,
"Cannot write the record since the file is not properly sorted.");
return(false);
}
if(myRefPtr != NULL)
{
record.setReference(myRefPtr);
}
// File is open for writing and the header has been written, so write the
// record.
myStatus = myInterfacePtr->writeRecord(myFilePtr, header, record,
myWriteTranslation);
if(myStatus == SamStatus::SUCCESS)
{
// A record was successfully written, so increment the record count.
myRecordCount++;
return(true);
}
return(false);
}
bool SamFile::myHasHeader [protected] |
Flag to indicate if a header has been read/written - required before being able to read/write a record.
Definition at line 399 of file SamFile.h.
Referenced by ReadHeader(), ReadRecord(), resetFile(), WriteHeader(), and WriteRecord().
 1.7.6.1
1.7.6.1 
